16 Apr 2020
GC Diagnostic Skills II | Reduced Peak Size
We are frequently asked about issues with reduced peak size in Gas Chromatography and I’m guessing this is related to just how difficult this problem is to troubleshoot. This isn’t necessarily related to the complexity of the problem and more to do with the number of possible causes. There are so many potential causes that an inexperienced Gas Chromatography (GC) user may not know where to begin the troubleshooting process.
Fear not, what follows in our logical guide to locating and fixing the issues with loss of sensitivity, and we’ve tried to cover as many of the instrument and application issues that we can think of.
Step 1 – Define the Problem
Sensitivity problems can be grouped by way of their presentation within the chromatogram;
- All peaks sizes (heights and or areas) decrease - retention times do not change
- All peaks sizes (heights and or areas) decrease - retention times shift, no evidence of loss of efficiency (peak broadening)
- All peaks sizes (heights and or areas) decrease – peaks are broadened, retention time shift may occur
- Early eluting peak sizes are reduced, later eluting peaks remain constant
- Later eluting peaks appear smaller, earlier peak sizes remain constant
- Specific peaks within the chromatogram are smaller, whilst others remain constant (or become relatively larger)
We should attempt to identify which of the above categories our problem falls into as this directly influences the sources of the potential problem, and consequently the actions we take to investigate and solve the problem.
All peaks sizes (heights and or areas) decrease - retention times do not change

Figure 1: Reduction in peak area for all analytes in a chromatogram, retention time and peak width remain constant.
As the description of this problem suggests, whatever the issue, it seems to be effecting all analytes equally, which leads us to think about physical problems with the instrument settings (including the autosampler) rather than thinking about chemical issues.
As always, it’s best to start with the most obvious possible causes as they are typically easier to check, verify and correct;
a) If operating in split mode check the inlet split ratio in the acquisition method and correct if required
b) If splitless mode with pressure pulse – check the inlet pulse pressure and duration in the acquisition method
c) Check that the Inlet and Detector Temperatures have been set correctly within the acquisition method
d) Check that the sample vial contains sufficient liquid and use separate vials containing the same sample liquid for repeated injections to rule out the possibility of sample loss through a compromised septum (especially when analytes are highly volatile) and to confirm that the problem is reproducible
e) Check that the autosampler syringe plunger is free within the barrel and does not leak, observe an injection cycle and confirm that sample is being aspirated from the vial and that it the correct volume (easier if the sample syringe is graduated)
f) Check and replace inlet septum as required, as well as checking that the correct liner has been installed (including liner geometry and any packing materials)
g) Check that the sample preparation / dilutions are correct as per the sample preparation method
h) With flame based or ionising detectors, check that the fuel gas ratios are appropriate and that all flow rates are as they should be (using a flow meter, checking one gas flow at a time), as well as checking all applied voltages according to the manufacturers specification and instrument acquisition method
i) With MS detectors in Selected Ion mode, check the masses and dwell times of each ion within each SIM group to ensure the cumulative ion count is matched to the acquisition method
j) With MS detectors in SCAN mode, check the MS tune and verify that the repeller or accelerator voltage has not increased dramatically (indicating a dirty ion source), the electron multiplier or MCP voltages have not increased dramatically (indicating a worn out detector) and that the ionisation energy is correctly set (typically 70eV for electron ionisation)
k) Check the detector attenuation range and set appropriately, as a secondary check, when the detector attenuation is incorrectly set, the signal to noise ratio for a given peak will not reduce, simply the chromatogram will appear ‘smaller’
All peaks sizes (heights and or areas) decrease - retention times shift, no evidence of efficiency loss (peak broadening)

Figure 2: Reduction in peak area of all chromatographic peaks with accompanying shift in retention time.
Where peak height or area reductions are accompanied by shifts in retention time but without noticeable reduction in efficiency, i.e. peaks do not broaden significantly, we begin to think about carrier gas flows and the column dimensions.
a) Check that the correct column dimensions have been entered into the data system and that the correct column is installed, paying attention to the stationary phase film thickness
b) Check that the carrier gas is flowing at the correct volumetric flow rate using a calibrated flow meter
c) Check that the carrier gas flow programming method is correct by selecting either constant pressure or constant flow operating modes. In the latter, the carrier gas pressure will be ramped to achieve constant linear velocity of carrier through the column as the oven temperature increases and the carrier gas viscosity reduces.
d) Change the inlet septum to overcome any leaks that may be occurring during the injection phase (when the sample syringe enters or leaves the inlet) or as column head pressure increases with the thermal gradient in constant flow operation
All peaks sizes (heights and or areas) decrease – peaks are broadened

Figure 3: Reduction in peak height or area with accompanying band broadening – note that peak apex retention time changes are also highly likely with this issue.
When all peak heights or areas reduce and the peaks broaden, the most obvious cause is a loss of efficiency within the chromatographic system. This phenomenon should also lead a reduction in signal to noise ratio for analyte peaks.
a) Check that the correct column has been installed and that the correct column dimensions and carrier gas flow rate have been set within the acquisition method
b) Check the column logs for column age and previous usage, if the column is old or has been used with dirty sample matrices, suspect that the column efficiency might have reduced.
c) Run a column test mix and compare with the result obtained originally
d) If loss of column efficiency is suspected, trim the column at the inlet end, typically 0.5-1 meters of column will need to be trimmed in order to properly restore performance
e) Verify that the column has been installed to the correct distance within the GC inlet and detector
f) For flame based and ionising detectors, check that the gas flows are correct paying particular attention to any make-up gas flow rates

Figure 4: Lower data sampling rate causing issues with reduced peak height and increased peak width.
As shown in Figure 4, incorrect detector settings may also cause a reduction in peak height, accompanied by peak broadening.
a) Check the detector acquisition rate to ensure it matches the settings within the acquisition method
b) If no data acquisition rate is specified within the method, experiment with different acquisition rate settings to obtain the optimum acquisition rate
c) If using MS detection, ensure that the detector settings are correct including a SIM group start and end times, ion dwell times and MS threshold settings. If using MRM detection, ensure that the correct MEM transitions have been set and the collision energy is correct
d) Check source parameter settings, especially the electron energy (typically set to 70 eV) and that the detector (electron multiplier or multi-channel plate) voltages are within the acceptable range (check manufacturers guidelines). Perform an MS autotune and compare to the previous tune report to ensure no major changes in applied voltages, which might imply a source clean is necessary or there is an air leak within the GC or MS modules.
Early eluting peak sizes are reduced, later eluting peaks remain constant

Figure 5: Injection of n-alkanes in which early eluting peaks are significantly reduced in height and area, which is accompanied by poor peak shape for the early eluting compounds. Top chromatogram starts with oven temperature too high to achieve solvent focussing, the problem is alleviated in the bottom chromatogram as the initial over temperature is lowered.
The reduction in peak height or area of early eluting peaks may point to some specific problems within the GC inlet or method acquisition conditions. For some of these problems, the reduction in peak size of the early eluting compounds is also accompanied by a deterioration in peak shape
a) Check and if necessary replace the inlet septum to overcome any sample losses during the injection phase as the sample syringe passes into and out of the GC inlet
b) Check the condition of the septum within the sample vial as more volatile analytes may be lost from the sample on standing if the vial lid has not been properly tightened, the septum is deformed within the lid or a previous injection from the vial has cause coring of the septum. Perform repeat injections of the same sample solution from different vials to test this theory.
c) In splitless injection, the reduction in peak height or area can be indicative of poor performance of analyte solvent focussing on the GC column and one should check all of the following factors;
i. Check that the correct sample solvent has been used for sample preparation
ii. Check that the initial oven temperature and hold time are correct within the acquisition method, if the initial column temperature is too high, this can lead to loss of analyte focussing within the column and where possible the initial column temperature should be 20oC below the boiling point of the sample solvent
iii. Check that the splitless time is correct within the acquisition method, too short a splitless time can often lead to the loss of early eluting sample components as they are vented via the split line
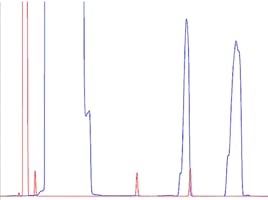
Later eluting peaks appear smaller, earlier peak sizes remain constant

Figure 6: Loss of later eluting compounds due to analyte discrimination with in the split/splitless inlet.
Again, the loss of sensitivity for later eluting peaks can lead to specific troubleshooting investigations which focus on the GC inlet and sample introduction processes. This phenomenon is known as sample discrimination and occurs due to the higher boiling nature of later eluting analytes.
a) Check that the syringe plunger (or injection speed) is correctly set. If the plunger moves too slowly through the syringe barrel, pre-volatilisation of the sample solvent may occur and higher boiling analytes may condense onto the inner surfaces of the syringe needle, never making it into the inlet. Fast injection speeds are preferred to ensure that the sample leaves the syringe in the liquid form.
b) Check that the correct liner has been used and importantly that any liner packing materials are of the correct material (deactivated glass wool or quartz wool are typical) and crucially are positioned in the correct place within the liner (usually so that the tip of the syringe needle just penetrates the packing or at the bottom of a lower restriction (goose neck) liner. The liner types should always be specified within the analytical method, however if it is not, then one may need to experiment with different liner types until the optimum liner geometry and packing materials are identified.
Specific peaks within the chromatogram are smaller, whilst others remain constant (or become relatively larger)
<
Figure 7: Loss of sensitivity with certain analytes due to GC system activity and adsorption of more polar analyte species.
When certain analytes (often initially appearing to be randomly spaced throughout the chromatogram or just one or two analyte peaks which are not closely eluting) then this symptom can appear very mysterious. Further, this problem can often be accompanied by a deterioration in peak shape of the affected peaks. When these symptoms occur, then out thoughts typically turn to matters of chemistry and to find a link between the analytes which have been affected. Typically, one may be able to establish a link such as a similar analyte chemistry and in particular more polar analytes are seen to be the linking factor. When polar analytes encounter absorptive groups (silanol groups from silica based materials or active metal surfaces) within the inlet liner or GC column, then analyte adsorption may occur, significantly reducing the number of analytes which reach the GC detector.
a) Typically one would trim a small length (0.2-0.5 meters) from the column inlet paying particular attention to the quality of the column cut and the correct positioning of the GC column within the inlet
b) Install a freshly deactivated or new inlet liner of the correct geometry and packing material
c) Perform inlet maintenance to change any consumable metal parts (bottom seals) and wash the inside surfaces of the inlet using a cotton swab dipped initially in hexane and then methanol
As I said in the introduction, a bewildering number of issues can lead to a reduction in instrument sensitivity when using GC or GC-MS instruments and it is important to identify the symptoms as described above in order to perform the correct troubleshooting checks and remedial actions. This guide should also help you to perform the troubleshooting investigations in the most efficient manner, starting with the most common and/or simplest checks initially, to save both time and effort in identifying and correcting the problems which have led to the reduction in peak height or area.
Check out the other instalments in this series:
GC Diagnostic Skills I | Peak Tailing
and
GC Diagnostic Skills III | Baseline Problems
Related articles