28 Aug 2017
Getting the best Repeatability from your Quantitative Gas Chromatography
I’m often asked ‘what reproducibility should I expect to get from my [insert instrument manufacturer and model]’? So most folks are referring to the repeatability aspects of precision, i.e. what relative standard deviation (usually expressed at %RSD) for peak area or quantitative result should I be able to achieve from repeat injections from a single vial of sample.
It seems the end user wants me to comment on the quality of the instrument and sees this as the determining factor in the repeatability of a particular analysis. Whilst instrument design and build quality are obviously important, it’s often the experimental variables, instrument set-up and equipment maintenance which have the most impact on repeatability of peak areas and quantitative data.
It should be possible to achieve <1% RSD for routine applications (assays for example) and between 2 and 5% RSD for trace analysis. Regulatory standards differ for various industries, analysis types and target analyte concentrations, however a figure of <2% RSD should be used as a guide when assessing repeatability with the notable exception of ultra-trace analysis and bioanalytical applications (i.e. analysis of physiological matrices). You should check your industry or company guidance to make sure you are not trying to achieve the impossible!
But to the business of this article – what can we do to ensure repeatability is as good as we can make it with quantitative gas chromatographic analysis?
There are many factors at play in a GC experiment which can affect repeatability, so I’ll work from the sample to the detector to make the workflow easier to follow.
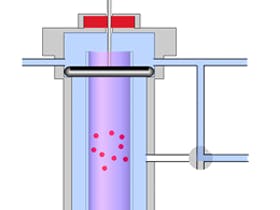
- Choose good quality vials and when filling, only fill to the shoulder of the vial, leaving a small headspace above the sample liquid. This prevents vacuum formation / cavitation when aspirating the sample from the vial which can lead to irreproducible sample volumes being drawn into the autosampler syringe. The quality of the glass used in vial manufacture can be important when dealing with adsorptive compounds and this should be assessed on an application by application basis.
- When making repeated injections from the same vial to assess quantitative repeatability, take into account the volatility of the sample solvent and the analyte. Once the vial septum is pierced there is a possibility for evaporation of either which can obviously affect the sample concentration. Some workers prefer to fill 6 separate vials with the same sample solution – filling each vial to the same level.
- The choice of sample solvent is very important due to the differences in expansion coefficient as the liquid sample is flash vaporised in the GC inlet. The amount of sample vapour created should be contained within the internal volume of the inlet liner and the volume of gas created will depend upon the expansion coefficient of the solvent, the inlet temperature and pressure (carrier flow). One can use a Vapour Volume calculator (http://agilent.com/en-us/support/gas-chromatography/gccalculators) to assess the possibility of overfilling the inlet and losing some analyte to the system under your operating conditions. Most 4mm i.d. liners will be capable of holding between 900 and 970 μL of vapour. A good rule of thumb is to use 1-2 μL of organic solvent and 0.5 μL if injecting water, but it’s better to check out the actuals using a calculator (see link above). Note here that water can sometimes cause repeatability issues with MS detectors due to large variations in source pressure as the solvent vapour passes through as well as issues with gas phase reactions with the ion source components which may cause changes in the source efficiency.
- Use an appropriate syringe volume. 5 μL and 1 μL syringes are preferred to 10 μL versions as they will give a more repeatable injection volume. Lower injection volumes should be matched with lower syringe volumes, so for an injection of 1 μL or less use a 1 μL syringe.
- Syringe washing is an often overlooked parameter and when optimised can significantly lower carry-over. You will need to experiment with your syringe washing and sample priming routines however I’ve previously found that 3-5 sample primes (sometimes called pumps) and three washes from two different solvent wash vials both before and after injection give reasonably good results. If the sample is particularly viscous, you may need to use a viscosity delay, to allow time for the viscous sample to rise up into the syringe and avoid variable volumes being injected.
- Check the syringe plunger speed. Where higher boiling analytes are involved, it’s very important to have a fast plunger speed to stop the sample volatilising inside the syringe needle which risks condensation of higher boiling analytes onto the syringe needle inner surfaces – which is a leading cause of sample discrimination.
- Ensure that the inlet is well maintained and this includes taking care to change any filters in the split line on a regular basis (check manufacturers recommendations) as well as giving the inlet internal surfaces a good clean with an intermediate polar solvent (ethyl acetate works well) every three to six months.
- Choose good quality septa which do not core and match these with an appropriate cone-tipped syringe needle to minimise the risk of coring which will cause pressure fluctuations during injection and risks sample loss which will affect repeatability. Further, where inlets have screw threaded closures, do not overtighten the inlet cap as this may lead to septa splitting, which will also risk sample loss during the injection phase. By the way – the same principles hold true for screw cap vials. As always, follow your manufacturer’s instructions.
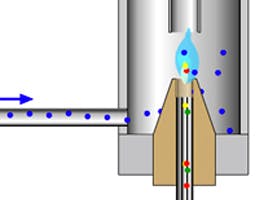
- Choose your liner carefully! Liners for split and splitless injections are typically of different design. Split injection liners may be ‘straight-through’ i.e. no baffles or packing or where higher boiling less volatile analytes are concerned, may contain a plug of deactivated glass wool positioned so that the tip of the syringe just penetrates into the packing. The packing serves to wipe any condensed analyte from the syringe tip, to provide a high surface area for less volatile analytes to condense and subsequently evaporate from within the liner and also to ensure good mixing of the sample vapour. For splitless injection the liner may contain a glass wool plug in the bottom of the liner as well as a baffle at the liner outlet – both of these features are designed to stop the analytes encountering the hot metal surface at the bottom of the inlet which may case adsorption or thermal degradation of labile analytes.
- Always use deactivated inlet liners which will give better repeatability especially with more polar analytes and inspect / change the liner regularly, especially where dirtier sample matrices are involved. Any liner contamination is a site for the potential adsorption of your sample and as such will affect quantitative repeatability. My recommendation is to buy good quality, deactivated liners of the appropriate design and to change them regularly.
- For splitless injection the initial oven temperature should be held for a period of time (30 seconds is typical but should be optimised) at 20oC below the boiling point of your sample solvent in order to ensure sharp analyte peaks and good quantitative repeatability. Further, the splitless time prior to the split valve being opened should also be optimised to ensure good solvent peak shape (solvent peak will tail badly if the splitless time is too long) and avoiding the loss of analyte through the split line (if the splitless time is too short). This is an empirically determined parameter and is often essential when performing quantitative splitless injection.
- The position of the column in the inlet and detector is critical to avoid problems with poor repeatability and manufacturer’s instructions on column installation should be closely followed. This is particularly pertinent for the column positioning in the inlet when performing split injection and if you haven’t accessed your manufactured instructions on this topic I would strongly urge you to do so.
- The cleanliness of the column in the first few centimetres and the quality of the column cut can also affect quantitative repeatability. If unsure about the provenance of the column, trim 10 – 20 cm from the inlet (ensure peaks are still within the i.d. windows which may have been set up within the data system) and ensure a good column cut (90o to the column wall and free from burrs), both of which will minimise the potential for analyte adsorption.
- Ensure the column is properly conditioned, again following manufacturer’s instructions, to minimise the impact of column bleed on sensitivity and repeatability.
- When using FID detection, ensure that the detector is clean, the column is positioned properly within the detector and there is a continuous (flow / pressure) supply of clean gas (free from oxygen and hydrocarbons – use gas filters where possible) in the correct stoichiometric ratio (typically 10:1:1 of Air / Fuel and Makeup).
- With MS detectors one should ensure that the instrument is properly tuned and meets specification limits.
- Finally, if after considering all of the above, you still have an issue, you may wish to consider the use of an internal standard to account for variations in the performance of the instrument. I urge you to consider this as a last resort, however if necessary, make sure you chose an internal standard which is available in high purity, with similar chemical and boiling point properties to the analyte / analytes of interest.
Phew – so much to consider in order to optimise the quantitative repeatability of our GC determinations, but all of these issues are worth careful consideration. My analogy is to that of an old bicycle – very often all the requisite parts are there, but it’s just a little old and creaky and makes you long for a shiny new one. However, with some care and attention it could be made to work as good as new. The same is true for chromatographic systems – there is rarely one catastrophic reason for poor repeatability, rather that there are several factors contributing to what is colloquially known as ‘system jitter’. Unless we pay close attention to all of the small things – then we may find ourselves with a sub-optimal performance.
Related articles