04 Dec 2017
HPLC Method Optimisation
It’s very easy to be comfortable with what you have. It’s only when we realise what could be, that we become interested in changing things.
Sure, we would all like to have a Ferrari California, but we know it’s out of reach and this limits our ambition, but what if we could aspire to something that is much better than our current vehicle and with a little effort could be a realisable ambition? An Audi R8 maybe or a Porsche Cayman?
I’ve outlined below the chromatographic conditions for the Chromatographic Purity USP monograph method for Lansoprazole;
Column: L1 150 x 4.6mm, 5μm
Injection: 40 μL
Detection: UV 285 nm
Flow Rate: 0.8 mL/min
Temperature: Ambient (controlled in this case to 35°C)
Mobile Phase (v/v): Solution A: 100% Water, Solution B: Acetonitrile, water, and triethylamine (160:40:1) adjusted to pH 7.0 with phosphoric acid.
| Time (mins) | Solution A (%) | Solution B (%) |
| 10 | 90 | 10 |
| 40 | 20 | 80 |
| 50 | 20 | 80 |
| 51 | 90 | 10 |
| 60 | 90 | 90 |
The remainder of the conditions can be found in USP Monograph: Lansoprazole, USP 40-NF 25.
I’ve modelled the separation obtained for Lansoprazole and 3 known impurities obtained on a 4 year old HPLC system with reasonably low extra column volumes and dwell volume (Figure 1).
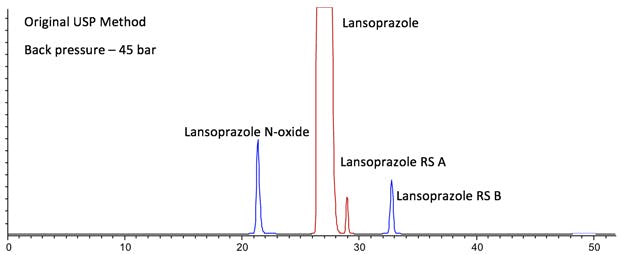
Figure 1: Computer model of Lansoprazole USP Chromatographic Purity separation.
One should note here that the relative responses should not be taken as accurate and that the tailing factor for the Lanzoprazole peak exceeded the required limits (TUSP = 1.5). We have increased the relative concentrations of each of the impurities for the purposes of this demonstration.
So – here we have our ‘current vehicle’ and we may be perfectly happy with its performance. But what might the separation look like if we ‘tuned’ it a little?
I would like to point out very clearly here that none of the following changes are acceptable under the current guidance of USP General Chapter <621>. But those who would like to see what might be theoretically possible please read on. For those who are not operating under legislative guidelines and who have the opportunity to improve methods, please also note that no changes should be made to methods without first validating the procedure according to your company guidelines.
The idea here is to keep within the operating pressure limits of a 400 bar to represent an ‘achievable aspiration’ in terms of the separation – i.e. one wouldn’t need a 1000bar ‘SuperSystem’ in order to achieve the proposed improvements.
To be honest, I’m also not really trying to improve the separation in any other respect than the time taken. How quickly could an acceptable separation be achieved?
In the initial set of experiments I’m going to follow a rule recommended by the USP (General Chapter <621>) regarding the limits over which one should alter the ratio of column length to particle diameter;
L/dp (-25% to +50%)
Although once again I should state that as this is a gradient separation, these changes are not allowable under current USP guidance. However, these adjustments would be allowable if the separation was performed under isocratic conditions.
Following the USP recommendations I’ve outlined in Table 1 the changes to column length and column dimensions that we may be able to consider. I’ve also targeted the use of superficially porous particles which will deliver higher efficiency than their fully porous counterparts and hopefully preserve the quality of our separations throughout the changes.
| Column Length (mm) | Particle Size (mm) | L/dp ratio |
| 150 |
5 |
30 |
| 100 | 4 | 25 (-17%) |
| 100 | 2.7 | 37 (+23%) |
Table 1: Changes to the column length to particle size ratio allowable for isocratic methods under USE General Chapter .
There is a further allowable change within the USP guidelines for isocratic methods of +/- 50% of the original flow rate, which might also be applied to our aspirational method. Note here again that some exceptions apply and that linear velocity should be maintained.
So lets look first at the separation obtained with scaled column dimensions and eluent low rate;
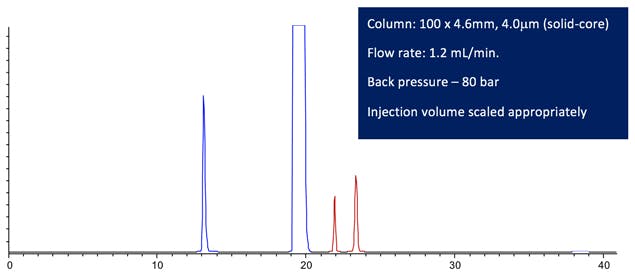
Figure 2: Computer modelled Lansoprazole chromatographic Purity Separation under the conditions stated in the figure. (All other conditions are maintained from the USP monograph method.)
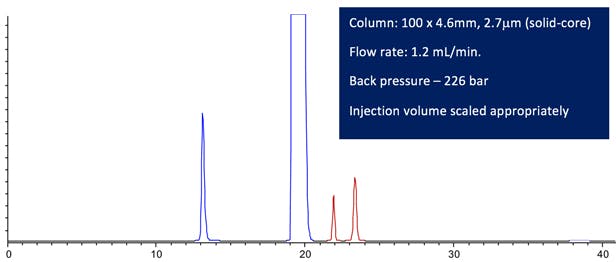
Figure 3: Computer modelled Lansoprazole chromatographic Purity Separation under the conditions stated in the figure. (All other conditions are maintained from the USP monograph method.)
The resolution between any critical pair satisfies the method requirements and the retention time of the last peak is around 75% of the original method. In fact the resolution is so good, one might consider further reductions in analysis time.
So, far I’ve followed the USP General Chapter <621> requirements for the method adjustments – but now I’m going to break free of these constraints and begin to look at optimizing the gradient conditions.
There is an excellent relationship that we might use to maintain gradient slope (and hopefully the resolution) whilst making method adjustments (Equation 1);
Gs = VmΔΦ/FtG
Where Gs is gradient slope, Vm the interstitial volume of eluent in the column, ΔΦ the gradient range (changing % organic), F is the flow rate and tG the gradient time.
Suppose I wanted to adjust the gradient time to 20 minutes but use the column specified in the original method.
Interstitial volume (Vm) = π x r2 x L x W
Where r is column radius (take care – not column diameter!), L is column length and W a porosity factor (0.68 can be used for fully porous particles and 0.55 for core-shell particles).
Interstitial volume (Vm) of USP method monograph (150 x 4.6mm) column is
π x 2.32 x 150 x 0.68 @ 1695(mL) @ 1.7mL
So from equation 1 and the monograph method we can calculate the gradient slope value.
Vm = 1.7
ΔΦ = 70 (gradient is 10 to 80% B)
F = 0.8
tG = 40
Gradient slope Gs (monograph method) = 1.7 x 70 / 0.8 x 40 = 3.72
So if we wanted to halve the gradient time we would need to retain a value of 3.72, and as we want to keep the column the same as the original method, we could either alter the gradient range (convenient but more likely to affect separation selectivity) or the flow rate, again convenient.
In order to retain a gradient slope value of 3.72 we would need to adjust the flow rate to 1.6 mL / min.
Gradient slope Gs (20 minute gradient, 150 x 4.6mm column) = 1.7 x 70 / 1.6 x 20 = 3.72
So lets see what the original monograph method would look like with a 20 minute gradient and a flow rate of 1.6 mL/min.
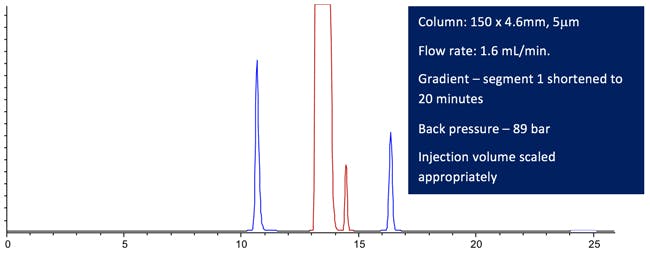
Figure 4: Computer modelled Lansoprazole chromatographic Purity Separation under the conditions stated in the figure. (All other conditions are maintained from the USP monograph method.)
So we’ve pretty much halved the run time and not suffered any ill effects from lost resolution.
Ok – so we might say that we’ve tuned up the current car a little. How aspirational might we be in terms of increasing method throughput? Could we combine the shortened gradient time and adjustments to column dimensions and flow rate to produce something a little more exciting in terms of run time but maintaining the separation quality and critical method attributes?
Using Equation 1, I’ve modelled several separations using various column dimensions, in each case altering the method flow rate to maintain, as closely as possible, the gradient slope value for the 20 minute gradient time. In each case I’m trying to maintain the usability of the method on a 400bar HPLC system.
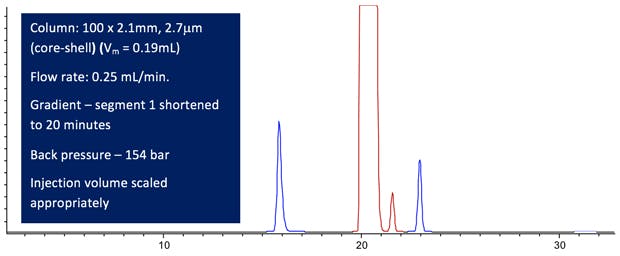
Figure 5: Computer modelled Lansoprazole chromatographic Purity Separation under the conditions stated in the figure. (All other conditions are maintained from the USP monograph method.)
From Figure 5 we can see we’ve gone backwards a little! Reducing the column volume (especially the column internal diameter) has dictated that we need to significantly drop the flow rate in order to retain a constant gradient slope and the method is slower than the previous iteration.
So – after a few more in-silico iterations – I threw all of my tuning tools at the method and came up with the following method by simply adjusting the various parameters in Equation 1 to maintain the gradient slope.
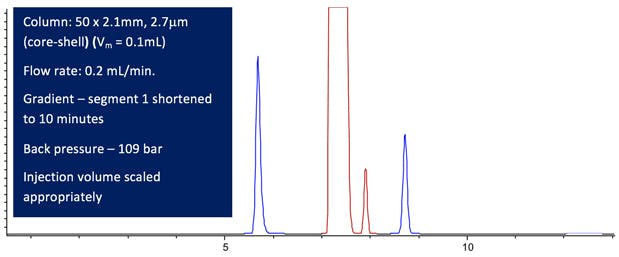
Figure 6: Computer modelled Lansoprazole chromatographic Purity Separation under the conditions stated in the figure. (All other conditions are maintained from the USP monograph method.)
A pretty successful ‘tune up’ I’d say with an impressive 75% reduction in analysis time.
But lets not get too carried away with our Audi A8 equivalent separation without first assessing our driving skills! What if we had a much older system with much larger dwell and extra column volumes.
Development System (used to this point) – Dwell Volume 600μL / Extra Column Volume <10μL
Older System (more realistic) - Dwell Volume 1100μL / Extra Column Volume 25μL
Our supertuned method above may look something like this;
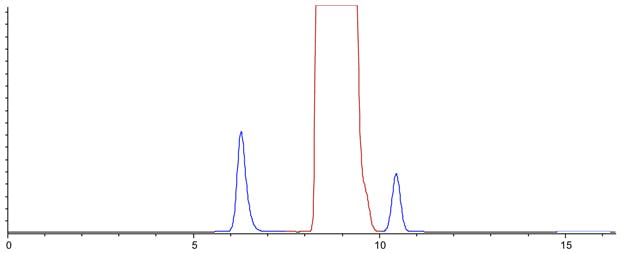
Figure 7: Computer modelled Lansoprazole chromatographic Purity Separation under the conditions stated in Figure 6 using a system with more realistic Dwell and Extra Column Volumes (All other conditions are maintained from the USP monograph method.)
Figure 7 shows us that this separation is clearly not acceptable! Our aspirations have overleapt our abilities….
So let’s back off a little and drive within the limitation may be our ability. This required some more modelling using the older system, however were still able to obtain a separation which reduced the overall run time by over 60%. The main changes are in the gradient time and crucially in the internal diameter of the column, which at 4.6mm is much more ‘forgiving’ in terms of the effects of larger extra column volume.
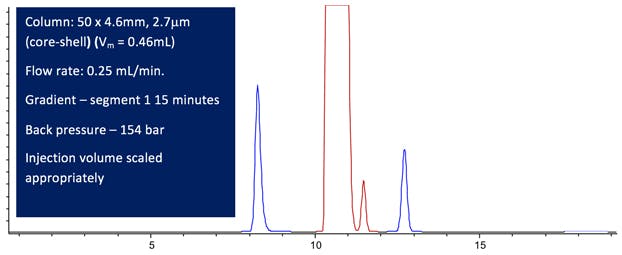
Figure 8: Computer modelled Lansoprazole chromatographic Purity Separation under the conditions stated in the Figure using a system with more realistic Dwell and Extra Column Volumes (All other conditions are maintained from the USP monograph method.)
Figure 8 shows us that the separation is satisfactory in terms of resolution and has a 60% run time compared to the original USP monograph method.
So what has been the point of this exercise?
- To highlight a bad monograph method? Not at all. Although I know this method is currently under review as there are other issues associated with it, from a chromatographic perspective it is fine, but dated and therefore longer than it needs to be.
- To annoy all of those who would like to improve their methods but can’t due to legislative restrictions? No, I merely want to highlight what is possible and to give a more aspirational perspective on the use of more modern column and instrument technology.
- To encourage anyone to break the rules laid out by the USP? Absolutely not, you should never do this, however once in a while it’s good to look up from our work and get a sense of what may be possible in the future, as the monograph methods catch up with newer technology.
For anyone using USP and other pharmacopeia methods, be aware that some changes to methods are possible and you should become familiar with the types of change that may be possible (using the appropriate Pharmacopeia) in order to improve a method or meet system suitability criteria.
For anyone laboring with methods using older column technology and who are fortunate enough to be able to make method changes, take the opportunity once in a while to ‘tune’ your methods or perhaps even to see what life might be like in the ‘aspirational supercar’.
Related articles