

03 Dec 2018
HPLC UV detection
UV detectors are undoubtedly the most frequently used type of detector with HPLC systems, and I predict this will continue for many years, despite the rise of the modular mass spectrometric detector.
I’ve written several times on the operational principles and variables used in UV detection, but I also get asked questions on the basic theory of UV detection and this time I’m going to address some of those FAQ’s to give a little more background theory as well as some operational tips and tricks.
Q1. Why are UV spectra so featureless – it makes it difficult to know which wavelength to monitor?
Some organic molecules can absorb electromagnetic radiation in the form of photons of UV and Visible light. Typically the wavelength range used in UV detection for HPLC is in the range 200 – 400nm, which covers both UV and the lower part of the visible spectrum. Figure 1 shows the electromagnetic spectrum and the relationship between the wavelength of light and frequency. It should be noted that the shorter the wavelength the higher the energy of the photons of light as defined by equation 1.
E = hc / λ
where E is energy, his Plancks constant, ci s velocity and λ is wavelength.
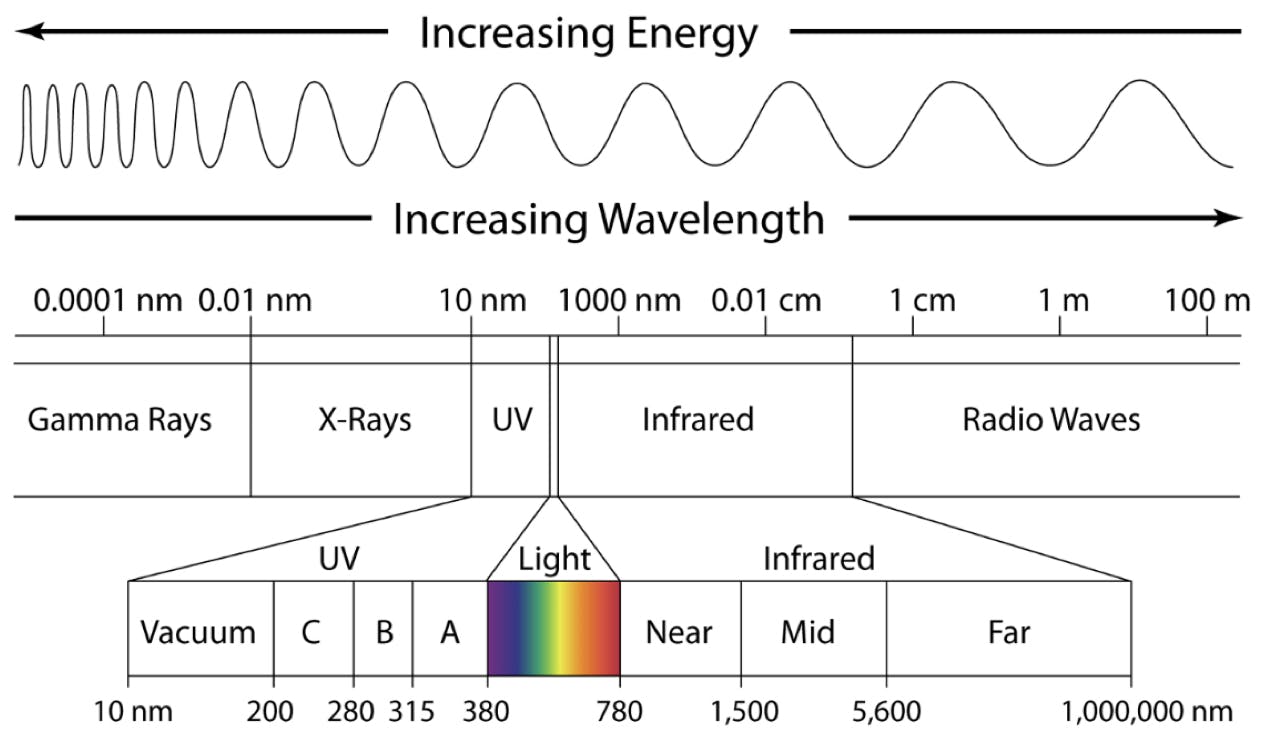
Figure 1: The electromagnetic spectrum and the relationship between wavelength and photon energy.
In order to absorb light energy, electrons within the atoms must be promoted from a ground state to an excited state and the specific energy transition will be dictated by the energy levels available, defined by the type of atoms and bonding with in the molecule, and the energy of the incumbent radiation, defined by the wavelength of light chosen.
Figure 2 shows some typical energy transitions resulting from absorption of UV and Visible light, when either bonding non-bonding electrons are promoted to higher energy (antibonding) orbitals.
![]()
Figure 2: Electronic transitions between various electronic ground states and excited states.
These various energy transitions can be roughly mapped against features of the organic molecules which are separated by HPLC.
| Molecule | Transition | λmax (nm) |
| Ethane | ||
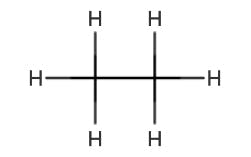 |
σ – σ* | 135 |
| Methanol | ||
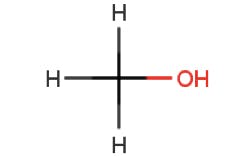 |
σ – σ* n – σ* |
150 183 |
| Ethene | ||
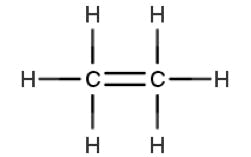 |
π – π* | 175 |
| Benzene | ||
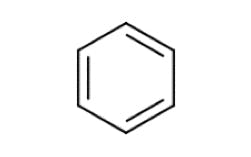 |
π – π* | 254 |
| Acetone | ||
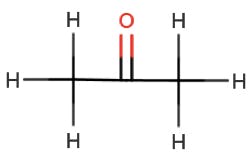 |
n – π* | 290 |
Table 1: Some typical electronic transitions and their corresponding λmax values.
So in theory, we can quite easily map the energy, and therefore the wavelength associated with the transitions caused when molecules absorb photons of light. However, is only part of the story and in reality, each of the orbitals into which the electrons are promoted have a number of sub levels associated with various vibrational and rotational states, any of which can be occupied by the promoted electrons, creating not just one, well defined transition, but many different possible transitions. Therefore, a transition that might be expected to yield a single well-defined signal, is actually ‘blurred’ over a number of energies or, as we observe in the UV spectrum, wavelengths. Figure 3 shows this concept in more detail.
![]()
Figure 3: Relationship between nominal electronic transition and various vibrational and rotational sub-levels which lead to broader spectra than expected in UV spectroscopy.
Whilst selecting the λmax from the spectrum in Figure 3 may be more obvious, it is true in general that the rather featureless spectra obtained from UV instruments make the selection of λmax and indeed the use of libraries for compound identification more difficult. However, read on for more details on how these problems may, to some extent, be overcome.
Q2. I’ve been told not to select the λmax for quantitative UV measurements due to robustness issues. Is this true?
In older UV instruments the stability and accuracy of the wavelength calibration was not as accurate as with modern instruments and therefore any small deviations in wavelength accuracy (drift) would lead to large change in the signal intensity, which would obviously be an issue. In modern instruments this need not be an issue – although a close eye should be kept on wavelength calibration and regularly checking response of a ‘calibrant’ is always a good idea. Be mindful also that most modern instruments will ‘self-calibrate’ as part of the instrument initiation routine – so it’s a good idea to power the instrument on and off from time to time!
All of this being said – there are other reasons why the λmax of the absorbance spectrum may change, for example when the solvent, additive or pH of the eluent change and this will lead to deviations in signal intensity. See Question 4 for more details on these topics.
Q3. Why do I sometimes need to choose low wavelengths and why does my baseline look noisy at lower wavelengths?
Electronic transitions involving ground state s electrons or non-bonding (n) electrons into their various antibonding orbitals usually require a large amount of energy and therefore typically occur in the 180-240nm region of the UV spectrum and are also of lower intensity compared to other transitions. For this reason, compounds which do not contain p electron systems (double bonds) typically occur at lower wavelengths) higher energies and this can be quite problematic when choosing UV detection wavelengths as the solvents and additive we use, can often interfere with the analyte absorption, so lowering sensitivity and adding to the general background noise. This being said, individual π-π* transitions involving just one double bond may also require higher energies and therefore may occur at lower wavelengths (e.g. ethene has a λmax of 175nm). Shifts of the λmax to higher wavelengths, which are less susceptible to interference from solvents or additives, typically only occur when the system is conjugated with multiple double bonds or are combined with other chromophores such as atoms containing lone pairs (n electrons) which are capable of electronic excitation (Figure 4).
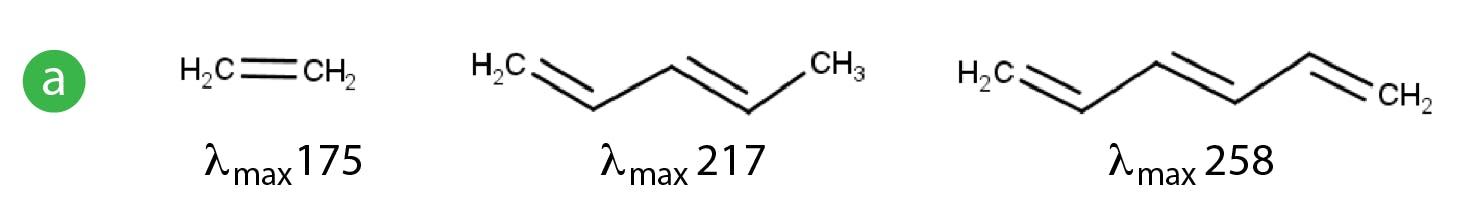
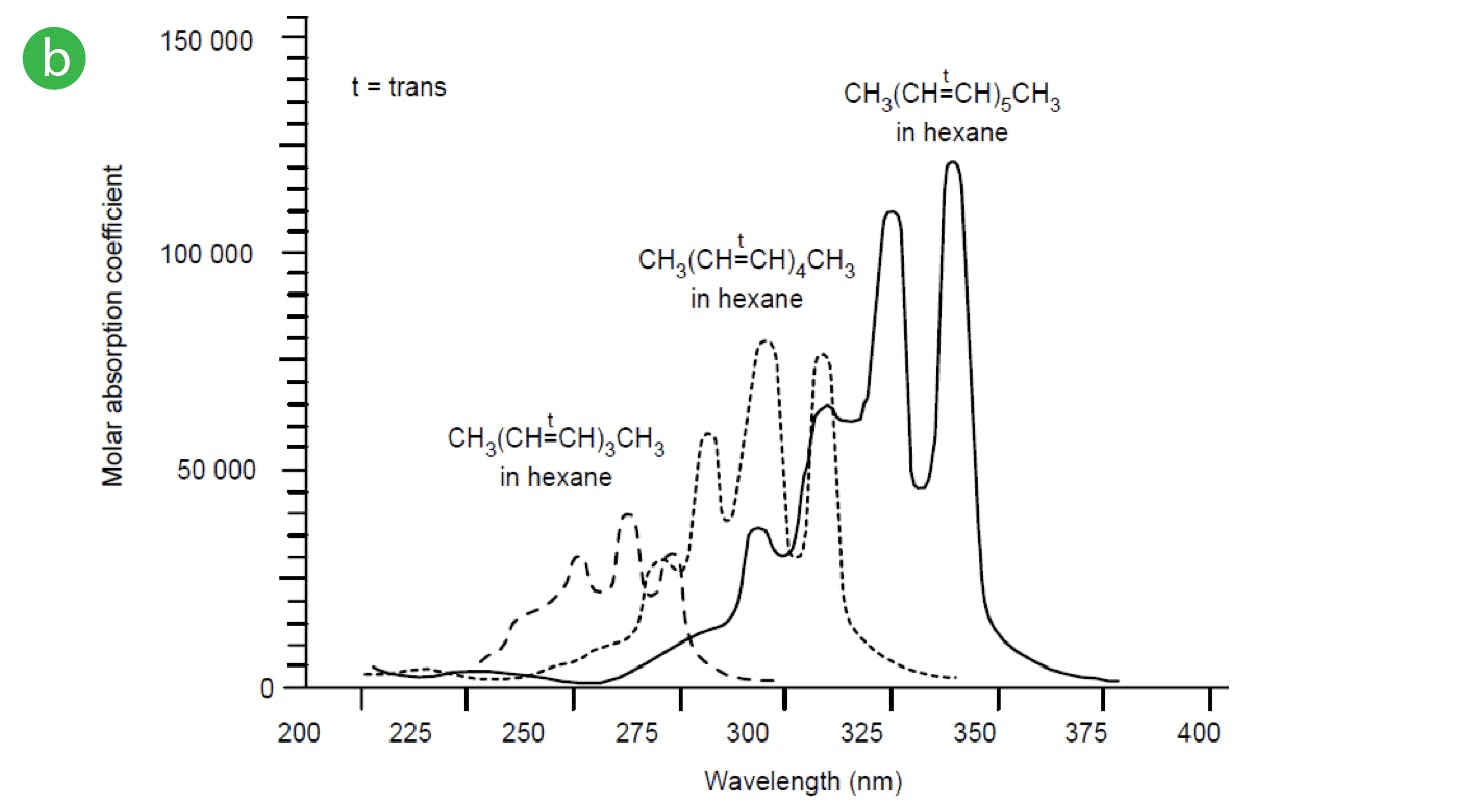
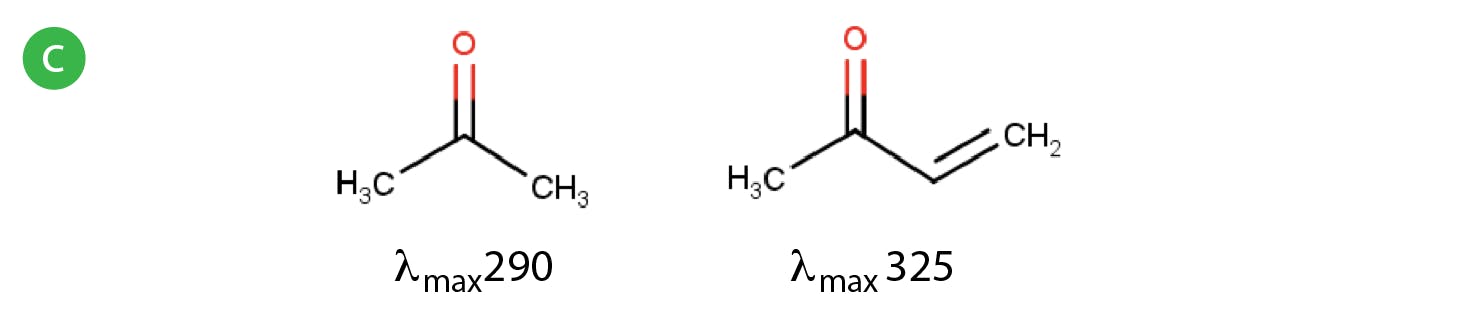
Figure 4: Shifts in UV spectra due to the effects of conjugation a) and b) and to the combination of molecular features.
It’s clear the absorbance characteristics are governed by the structural features of the whole molecule rather than individual chromophores, especially when there are multiple groups capable of electronic excitation via photon energy. An auxochrome a substituent that increases absorption of a molecule, typically methyl, hydroxyl, alkoxyl or amino group or a halogen atom; when the auxochrome is conjugated with a π-electron system, the λmax value is shifted to a longer wavelength.
Whenever a shift to a higher wavelength occurs, this is known as a red shift or a bathochromic shift. If a shift occurs to a lower wavelength, this is known as a blue shift or hypsochromic shift.
All of the above being said, in order to evaluate analyte for its suitability for UV detection, its always good to have an idea about the important chromophores typically encountered in organic molecules. The following links contain a host of information on the UV absorbance characteristics of functional groups and structural moieties;
UV-visible Absorption Maxima of Chromophores
Table 2 shows solvent UV Cut-Off values for some common HPLC solvents and additives. UV cut off is defined as the wavelength at which the pure component has an absorbance of 1Absorbance Unit (AU) in a 1 cm pathlength cell with water as reference.
| Solvent / Additive | UV-Cut off (nm) |
| Acetonitrile | 190 |
| Water | 190 |
| Methanol | 205 |
| Trifluoroacetic Acid (TFA) | 210 |
| Phosphate buffer (pH 2-3) | 210 |
| Diethylamine | 210 |
| Formic Acid | 210 |
| TRIS | 225 |
| Acetic Acid | 230 |
| Triethylamine | 235 |
| Acetate Buffer (pH 3.5 – 5.5) | 240 |
| Citrate Buffer | 250 |
| DMF | 268 |
Table 2: UV Cut-Off values for some typical HPLC solvents and additives.
Q4. What effects do different solvents / buffers have on UV sensitivity?
This is a very interesting question and is often misunderstood or ignored by users. There are many circumstances under which spectrum, and therefore the λmax value, may undergo bathochromic or hypsochromic shifts, resulting in a change in signal intensity if a single wavelength is being monitored.
It is fair to say that while all measurements are taken under the same eluent conditions, then quantitative measurements will be valid, however what if the eluent composition changes slightly during the analysis? Further, what may have been a good wavelength in a certain eluent, may not be so when the eluent is changed; this can be particularly relevant when undertaking eluent screening / optimising during method development.
The polarity of a solvent can modify the electronic environment of the absorbing chromophore. This can change the absorbance maxima and ε values for a molecule. For comparative analysis, a single solvent should be used for all measurements.
The Frank-Condon principle states that during an electronic transition the atoms do not move; whereas electrons, including those of the solvent molecules, will reorganize during an electronic transition. Most transitions result in an excited state which is more polar than the ground state.
Therefore, polar solvents will be able to interact to a greater extent with the more polar excited state via dipole-dipole interactions. These stabilizing interactions will lower the energy of the excited state but will have a negligible effect on that of the ground state, resulting in a change of absorbance maximum.
For example, it has been seen that ethanol (polar) solutions produce spectra with longer wavelength maxima in comparison to hexane (non-polar) solutions. A small red shift of approximately 10 -20 nm (π-π*) is observed when the solvent is changed from hexane to ethanol.
For n → π* transitions a solvent effect is seen in the opposite direction (producing blue shift). The shift observed in the wavelength is attributed to a reduction in solvent hydrogen bonding in the excited state.
Carbonyl groups in particular can hydrogen-bond to solvents, which can accentuate the change to the UV spectrum when measured in polar solvents. For example, changing from hexane to water as the solvent for propanone changes the absorption maximum from 280 to 257 nm (Figure 5).
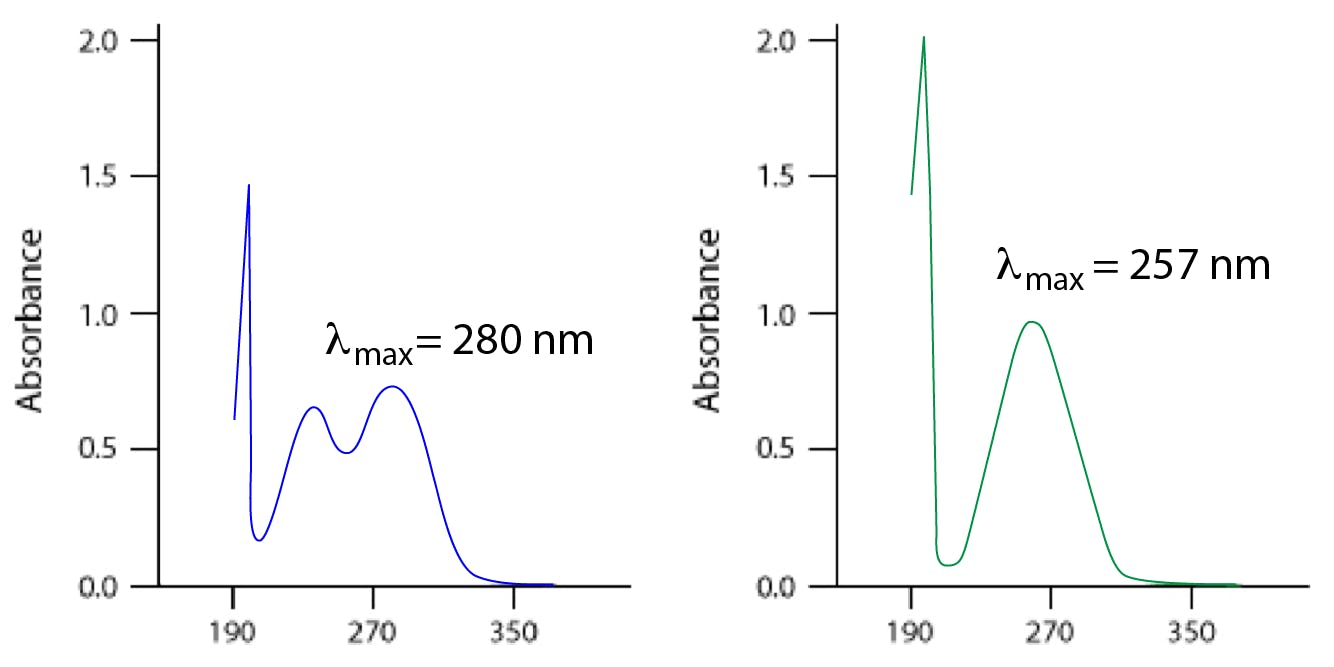
Figure 5: UV-visible spectra of propanone in hexane (left) and propanone in water (right).
Effects of pH on absorbance spectra can be very large and result primarily from the shifting of equilibrium between two different molecular forms, which may include from ionised to non-ionised or vice versa, as well as analyte conformational or shape changes.
Buffers can help control pH but remember that they also may absorb UV at the intended wavelength and therefore sensitivity may be reduced / noise increased.
One needs to be particularly mindful of pH changes over extended analytical campaigns when using volatile buffers or pH adjusting reagents as the volatile component is lost into the eluent reservoir headspace over extended periods of time.
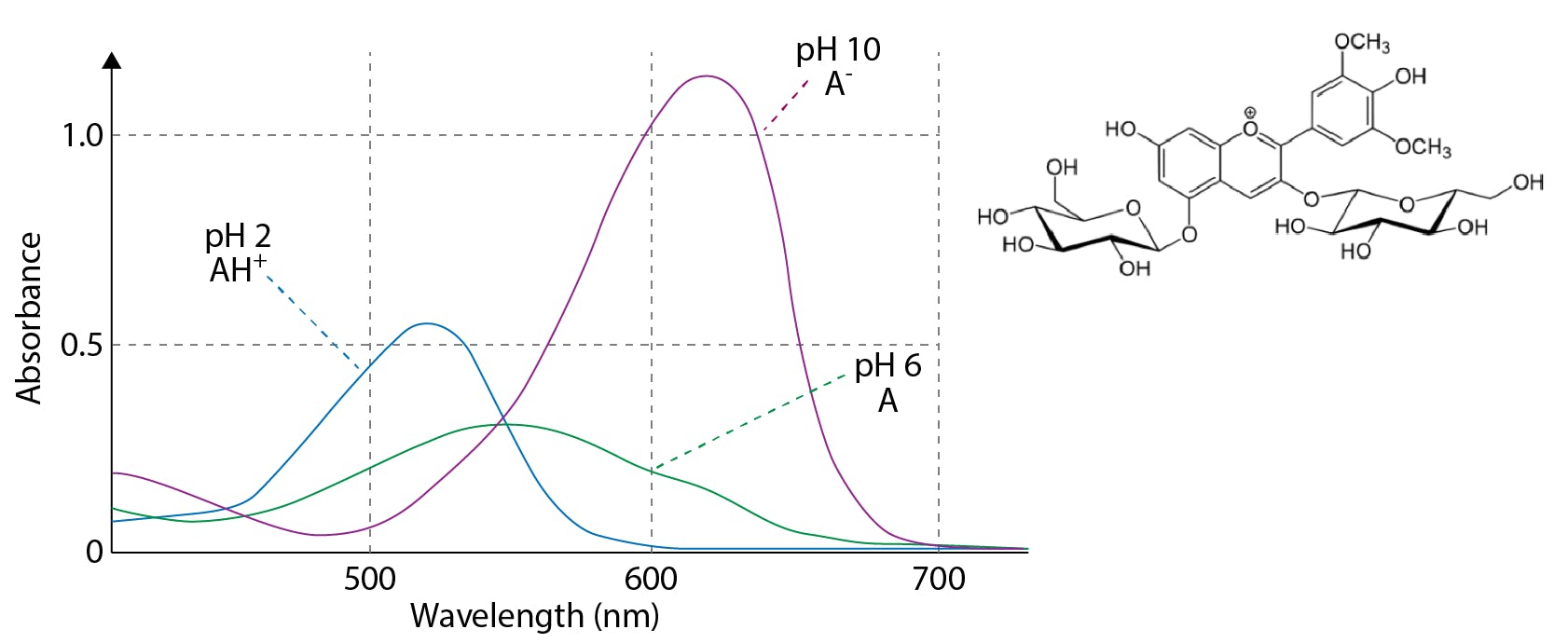
Figure 6: Anthocyanin pigment in buffers of varying pH.
Temperature can also have a pronounced effect on UV spectra, and these possible variations due to changes to the HPLC column oven temperature also need to be borne in mind when making changes to HPLC methods.
Q5. Can I really use UV detection to track peaks when developing methods or screening different columns or mobile phases?
As we discussed in Question 1, solution-based UV spectra for many organic compounds can be rather featureless, unlike vapour phase spectra which can be feature rich. Further, the amount of detail within the spectra can be dependent upon the composition of the eluent in which they are measured as shown in Figure 7.
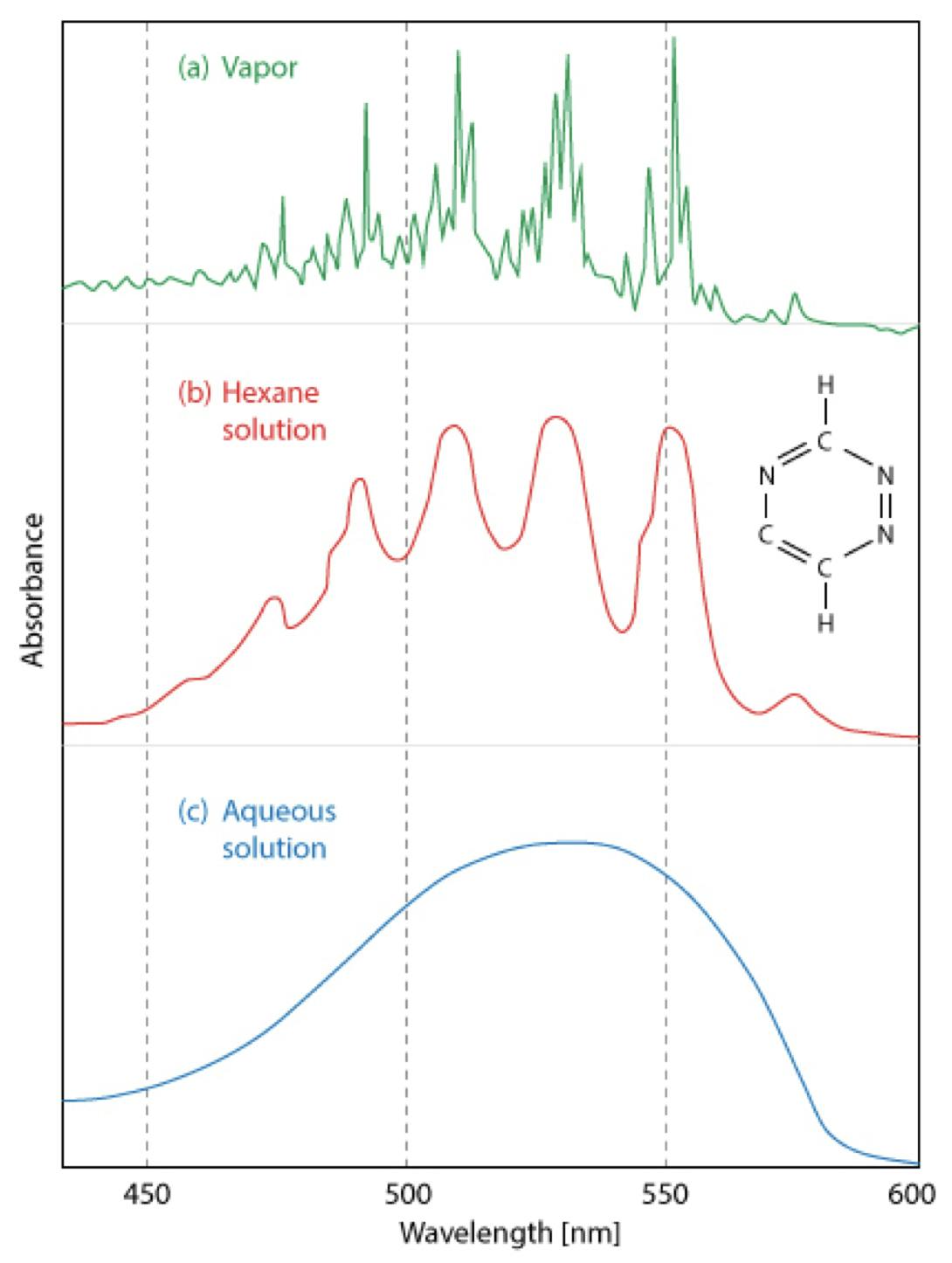
Figure 7: Comparison of UV-visible spectra of 1,2,4-triazine in the vapor phase, hexane, and aqueous solution.
However, even when 0th order spectra are rather featureless, which makes the use of libraries and peak tracking rather difficult, all is not lost.
Most UV detector or CDS software can create ‘derivative’ spectra (dA/dλ) and these spectra contain increasing greater numbers of useable maxima and minima which add data points to the spectra which can be very useful for increasing confidence when identifying or tracking peaks (Figure 8).
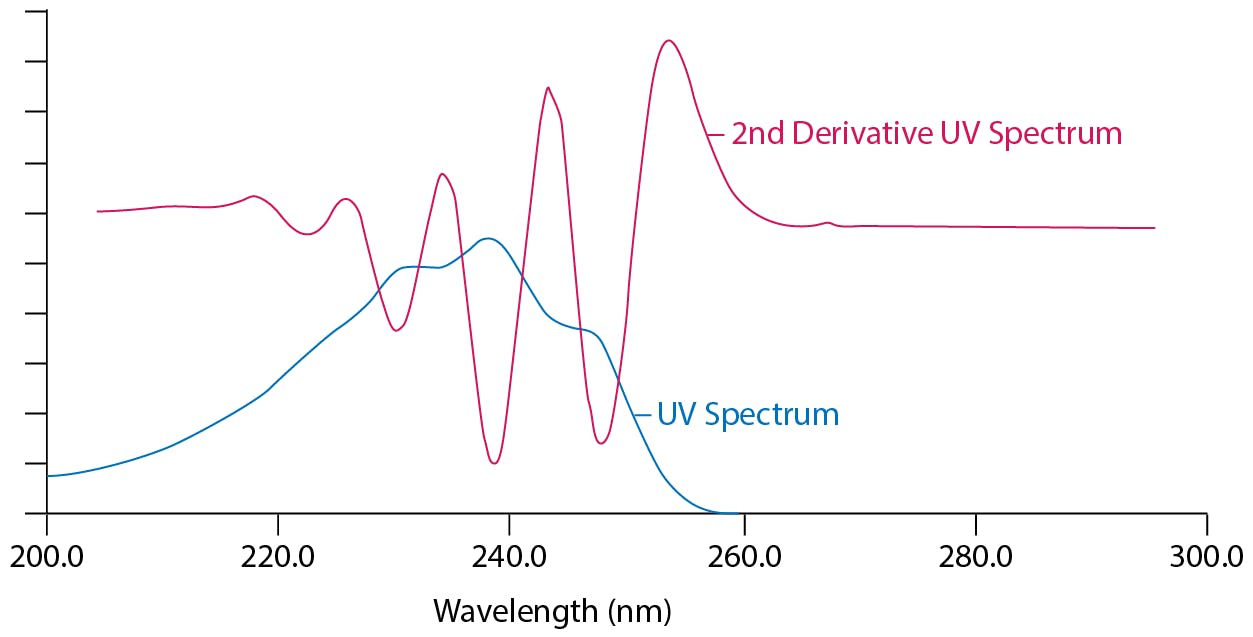
Figure 8: UV and second derivative UV spectrum of simvastatin (reproduced from Ref: 1)
It should be noted that higher order derivatives (2nd, 3rd etc) show a marked decrease in sensitivity as data is lost during the mathematical transformation and therefore 1st order derivatives should be chosen where possible. Further, there needs to be sufficient data captured during acquisition in order for the transformations to be meaningful.
Check your CDS supplier or manufacturer for instructions on how to achieve the data transformation and add derivative spectral to libraries.
References:
[1] Second-derivative UV spectrometric determination of simvastatin in its tablet dosage form, Lei Wang, Mandana Asgharnejad, Journal of Pharmaceutical and Biomedical Analysis, January 2000
Related articles




