02 Oct 2017
Retention Shifts in HPLC
Retention Shifts in HPLC
The nature of retention time changes in HPLC tends to fall into categories. Firstly, the retention time may ‘drift’ over several injections or several analytical campaigns and secondly, the retention time may suddenly ‘jump’ to a different value between injections or between analytical campaigns (i.e. analyte retention times are very different to when that method was run last).
Here are just a few of reasons why these symptoms might occur.
Start with the obvious. Ok so retention time ‘jumps’ may be due to errors that you made when setting up the method. For example, the wrong mobile phase flow rate may have been selected, the wrong column hooked up to the system or the wrong column oven setting made. So, whenever investigating drastic shifts in retention, start with the obvious and check your method settings.
Of course, there are other more insidious method problems which may also need investigating. One of the most common is large retention shifts when running gradient elution methods and moving between different instruments. Most probably the instruments will have different dwell volumes (the time taken for a change in eluent composition to be delivered from the pump to the analytical column). If these differences are significant and no account is made for them by altering the isocratic section at the beginning of the gradient or a ‘pre-injection’ is made – then significant retention differences will occur between instruments. Also note that resolution or selectivity differences may also occur under the same circumstances. Whilst we are talking about gradients – not all pumps like mixing gradients where one or other component goes below 5%. A little too long to explain here – however several phenomena are involved including de-wetting and cavitation.
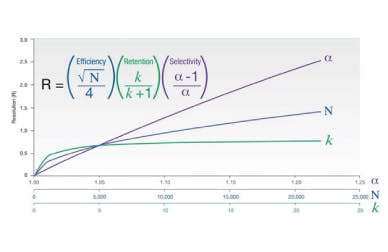
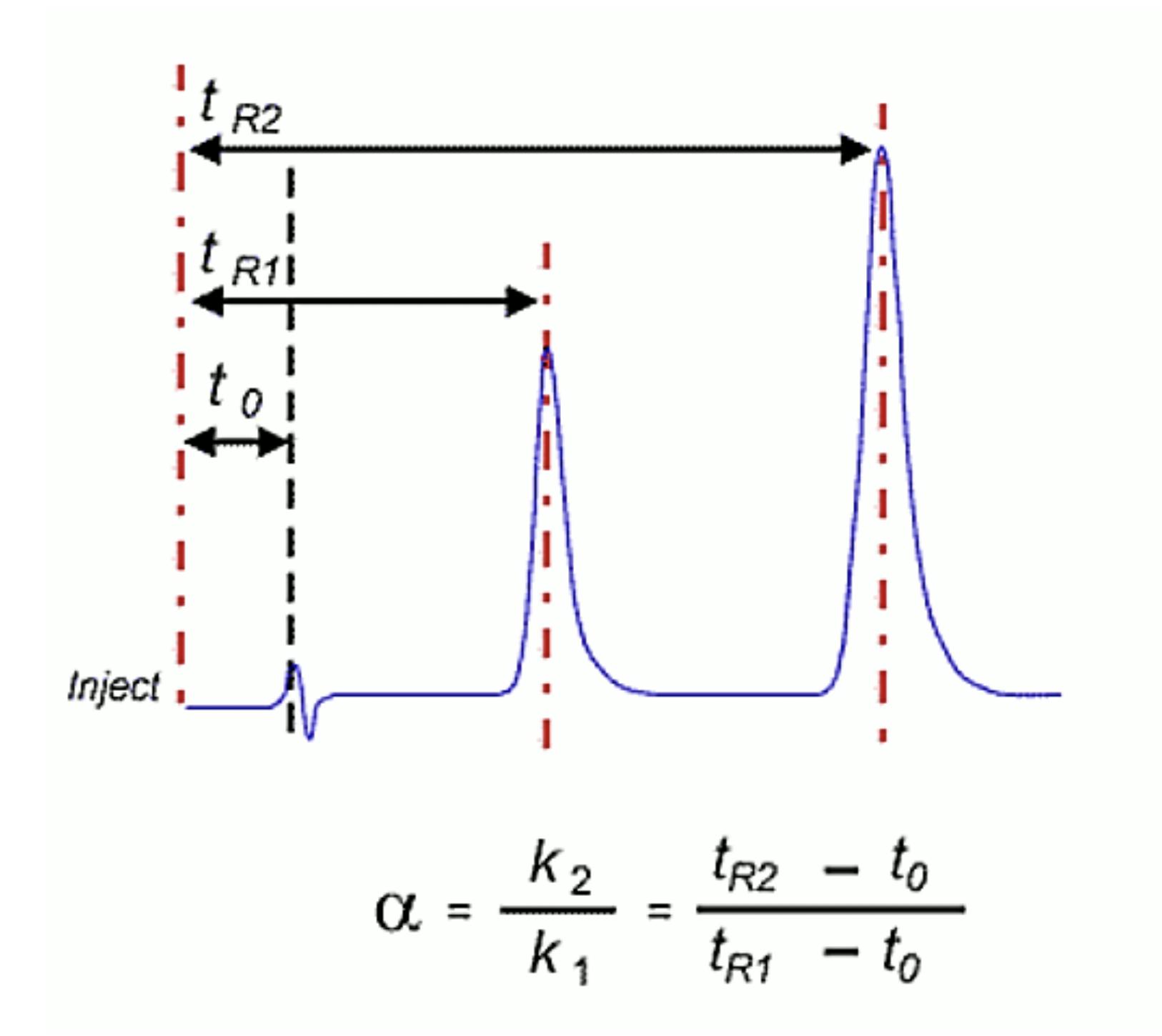
One should also probably check the function of the pump and that it is delivering at the volumetric follow rate set within the method. One can simply differentiate between hardware and chemical problems with retention time issues. Use retention factor as a marker – if retention factor remains unchanged but retention time shifts appreciably then the hardware is generally at fault – i.e. a flow rate change is affecting the elution time of the unretained t0 marker as well as that of the analyte. If the elution time of the unretained material changes relative to that of the analyte then the retention factor will change and this points towards a ‘chemical’ problem within the system. Remember that retention factor is represented by the equation:
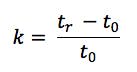
Where tr is the analyte retention time and t0 is the retention rime of an unretained component or the first baseline disturbance caused by the eluent passing through the detector.
There are some serious electronic flow meters available however I never really favoured these over the trusty 10mL measuring cylinder and the pump operating a 1mL/min. for 10 mins. You’ll soon get an idea if there is a problem. For added accuracy a 10 mL volumetric flak can also be used. One should carefully check for small leaks within the system – remember that ‘blue roll’ is fantastic for holding up to joints and watching the paper darken as the liquid you never noticed seeps into the adsorbent material.
Drastic retention time shifts can also be brought about by incorrectly prepared mobile phases. The wrong way to prepare a mobile phase could be the subject of a rather large book – however here are some of the common culprit...
- Don’t make up an isocratic mobile phase in a single measuring cylinder and especially don’t add organic to aqueous and make to volume – you will get the incorrect solvent ratio (lower amount of organic than expected) and your retention times will all shift to a later value.
- Make sure you have the correct buffer and are using it at the correct pH - remember that buffers are most effective at ± 1.0pH unit from their pKa. The buffer concentration should be capable of resisting changes in pH as the sample diluent and eluent mix within the autosampler and connecting tubing but most importantly at the head of the analytical column.
- Adjust pH of the aqueous portion of the eluent only – ensure the pH is properly calibrated.
- For pre-mixed mobile phases (organic and aqueous solvents already mixed) don’t degass ultrasonically or filter under vacuum as both result in loss of organic component (again retention times will be later than expected when operating in reversed phase mode)
- Don’t leave eluents standing on the instrument for too long – ingress of CO2 can substantially lower the pH of aqueous buffers
Drifting retention times can be a little more difficult to troubleshoot. One might start by considering the age and condition of the column being used. Are you seeing drift due to gradual loss of stationary phase (pH < 2.5) or dissolution of the silica matrix (pH > 7.5) in which case use columns designed to operate at extremes of pH. Of course – columns do ‘die’ naturally over time and drift under this circumstance is usually to shorter retention time and is accompanied by loss of efficiency (broader peaks).
Other typical issues include housing an non-thermostatted column on the open bench under the air conditioning unit. In modern times – no column should be seen without a jacket, a thermal jacket that is!
A changing eluent can also cause retention time drift. As the eluent reservoir empties the large headspace created in the bottle can accept a large amount of volatilised organic from a pre- mixed mobile phase and change the composition quite significantly. Always loosely cap the eluent jar (not with blue roll or lab film either – for reasons that we won’t go into here.
Developing leaks also tend to give rise to a drift in retention time. A thorough investigation of the hardware and connections with the trusty blue roll are recommended to hunt the leak.
When analysing ionisable analytes, ingress of CO2 on standing, as mentioned above, can lower the pH of the eluent - resulting usually in an increase in retention time for acidic species and a reduction in retention time for basic analytes.
There are more complex causes of retention time change - but I think this is probably enough for now and I hope you aren’t reeling from all of the issues you recognised in the text. By the way – don’t forget to check and change one thing at a time. I know we are all under major pressure to get results out of the door – but it makes sense in the long run!
Related articles