08 May 2018
USP Chapter 621 changes
In the December edition of this column, I wrote about Supercharging HPLC methods and used a USP method for Lanzoprazole to discuss how we might improve the method. One of the major caveats that I kept repeating in the article was that the changes we made to the gradient profile were outside of the ‘allowable changes’ mandated in the General Chapter of the USP in order to meet system suitability requirements (USP 40 - NF35 (Supplement 2) is the current version). This has been, in my opinion, a major limitation of the ‘allowable changes’ regulations to date.
Shortly after writing that December edition, a good friend who works in the Pharmaceutical Industry made me aware of a discussion document which was currently out for comment (C188676 (43(5) Harmonisation Stage 4 General Chapter 621), which contains proposed updates to the ‘allowable change’ regulations – and some of these are very interesting! For reference – I’ve tabulated the current regulations and those which are being proposed;
Current Guidelines
|
Variable |
Allowable changes |
Isocratic |
Gradient |
|
Column Chemistry |
Stationary phase may be changed within the same ‘L’ classification |
Yes |
Yes |
|
pH |
±0.2 |
Yes |
Yes |
|
Buffer Concentration |
±10% |
Yes |
Yes |
|
Mobile Phase |
The lesser of ±30% relative or ±10% absolute for minor components |
Yes |
Yes (but not recommended) |
|
UV Wavelength |
0, but error ±3 nm OK |
Yes |
No |
|
Column length and particle size |
L/dp = -25% to +50% or N = -25% to +50% |
Yes |
No |
|
Column diameter |
OK, if linear velocity is constant |
Yes |
No |
|
Flow rate |
OK if linear velocity is constant, plus an additional ±50%, with exceptions |
Yes |
No |
|
Gradient profile |
Changes to initial isocratic time of the gradient only |
- |
Yes |
|
Injection volume |
OK, if performance is OK |
Yes |
Yes |
|
Column temperature |
±10°C |
Yes |
Yes |
Recommended method for adjusting flow rate to maintain constant linear velocity;
![]()
Guidelines under Discussion
|
Variable |
Allowable changes |
Isocratic |
Gradient |
|
Column Chemistry |
No change of the physicochemical characteristics of the stationary phase permitted, i.e., chromatographic support, surface modification, and extent of chemical modification must be the same* |
Yes |
Yes |
|
pH |
±0.2 |
Yes |
Yes |
|
Buffer Concentration |
±10% |
Yes |
Yes |
|
Mobile Phase |
The lesser of ±30% relative or ±2% absolute for minor components |
Yes |
Yes (but not recommended) |
|
UV Wavelength |
0, but error ±3 nm OK |
Yes |
No |
|
Column length and particle size |
L/dp = -25% to +50% or N = -25% to +50% |
Yes |
No |
|
Column diameter |
OK, if linear velocity is constant |
Yes |
No |
|
Flow rate |
OK if linear velocity is constant, plus an additional ±50%, with exceptions |
Yes |
No |
|
Gradient profile |
Adjust each gradient segment according to Equation (1) below ** |
- |
Yes |
|
Injection volume |
OK, if performance is OK |
Yes |
Yes |
|
Column temperature |
±10°C |
Yes |
Yes |
Note * - a change from totally porous particle (TPP) columns to superficially porous particle (SPP) columns is allowed provided the other column chemistry requirements are met.
Recommended method for adjusting each gradient segment;
![]()
Note ** -
Composition/gradient: Adjustments of the composition of the mobile phase and the gradient are acceptable provided that:
- system suitability requirements are fulfilled
- principal peak(s) elute(s) within ±15% of the indicated retention time(s); this requirement does not apply when the column dimensions are changed
- final composition of the mobile phase is not weaker in elution power than the prescribed composition
So, as you can see there are proposed changes to the allowable adjustment to the eluent composition, further qualification of changes to the stationary phase as well as, critically, some guidance on allowable changes to the gradient profile.
I have to just re-iterate at this stage that these are merely proposals at the moment, they are not and may never be current. However, let’s assume that they are and discuss what this may mean in practice.
Starting with the proposed adjustments to the gradient profile, we can use the USP method for Lanzoprazole and it impurities, alongside an allowable change in column dimensions and flow rate to explore what an ‘adjusted’ method may look like.
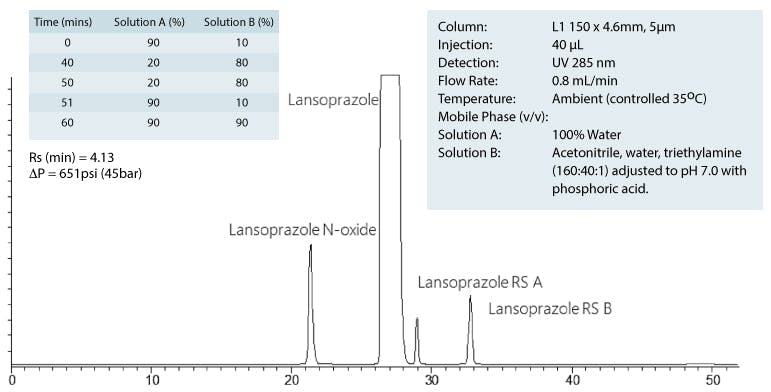
The USP method conditions for the analysis are;
| Column: | L1 150 x 4.6mm, 5μm |
| Injection: | 40 μL |
| Detection: | UV 285 nm |
| Flow Rate: | 0.8 mL/min |
| Temperature: | Ambient (controlled 35oC) |
| Mobile Phase (v/v) | |
| Solution A: | 100% Water |
| Solution B: | Acetonitrile, water, triethylamine (160:40:1) adjusted to pH 7.0 with phosphoric acid. |
Gradient Table;
|
Time (mins) |
Solution A (%) |
Solution B (%) |
|
0 |
90 |
10 |
|
40 |
20 |
80 |
|
50 |
20 |
80 |
|
51 |
90 |
10 |
|
60 |
90 |
90 |
We wish to translate the method to the following column - L1 50 x 2.1mm, 1.9mm and in this case the stationary phase will remain unchanged. This change is allowed under the current guidance as the L/dp ratio change is approx.. -12%
The flow rate translation in order to maintain constant linear velocity is;
![]()
To illustrate the changes to the gradient table – we have translated the first section of the original gradient;
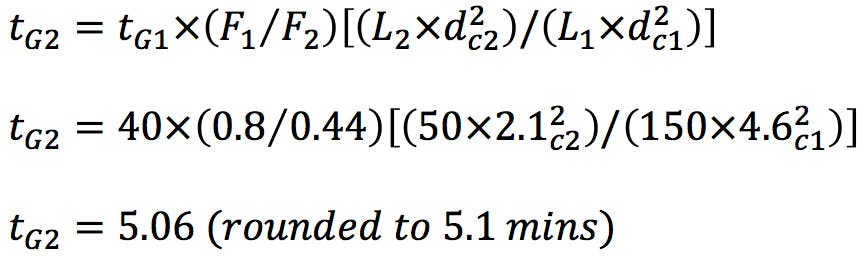
So the gradient table for the new method would be;
|
Time (mins) |
Solution A (%) |
Solution B (%) |
|
0 |
90 |
10 |
|
5.1 |
20 |
80 |
|
6.3 |
20 |
80 |
|
6.4 |
90 |
10 |
|
7.6 |
90 |
90 |
So the resulting chromatograms for the two methods would look as follows;
Original Method
New Method
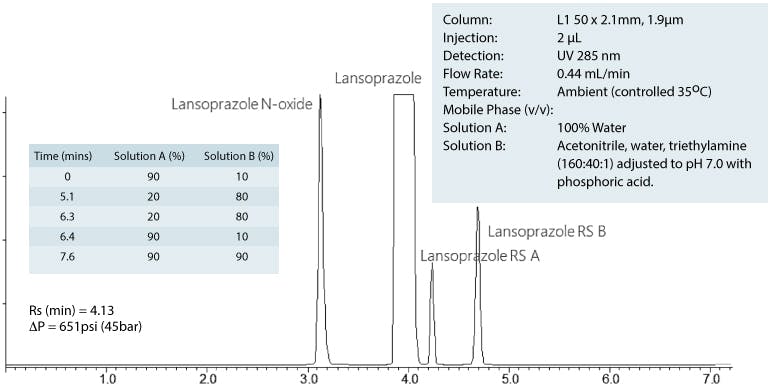
I think you would agree that, for those with 600Bar HPLC systems or better, the second chromatogram offers a much higher throughput option, whilst still complying with the proposed new guidelines.
Turning our attention to the other ‘major’ change which concerns some further clarification on allowable changes to stationary phases.
The guidance under consultation states;
No change of the physicochemical characteristics of the stationary phase permitted, i.e., chromatographic support, surface modification, and extent of chemical modification must be the same; a change from totally porous particle (TPP) columns to superficially porous particle (SPP) columns is allowed provided these requirements are met.
I think this is a little ambiguous to be useful as it stands.
What would constitute a change in the chromatographic support if swapping from a TPP to an SPP is allowed?
What now constitutes a change in surface modification? Does this refer to the bonded phase ligand, any endcapping reagents or surface area? If so – there will be little scope for changing from one manufacturers phase to another – even if they may be considered to be equivalent. If the ‘extent of chemical modification’ refers to the carbon load – this is a very crude measure of retentivity and in some cases, selectivity.
I absolutely applaud the wish to tighten the guidelines as, of the almost 200 commercially available columns in the L1 category, the chromatographic behaviour within this group can change drastically.
Perhaps this is the ideal time to place more focus on the selectivity databases which exist, which can help to demonstrate the ‘equivalence’ of selectivity of two different phases. At Element, we use a model based on the PQRI selectivity database (Hydrophobic Subtraction Model, HSM [1]), alongside some other key metrics to help clients understand the equivalence of reversed phase HPLC stationary phases. Alongside the HSM model we use a linear regression model for measuring the equivalence of each selectivity characteristic [2,3] and other checks to demonstrate the theoretical equivalence of columns and have found success with this model – some examples of reports and graphical comparisons are shown below for reference.
The Fs comparison number is the PQRI / HSM similarity factor, the Total Equivalence metric is based on a regression analysis of the differences in each HSM factor between the two columns, each reduced by the hydrophobicity and the Q comparison predicts the likelihood of critical resolution being maintained from the original separation. The model is built to take into account the presence and strength of acids and bases within the analyte mix as well as the eluent pH.
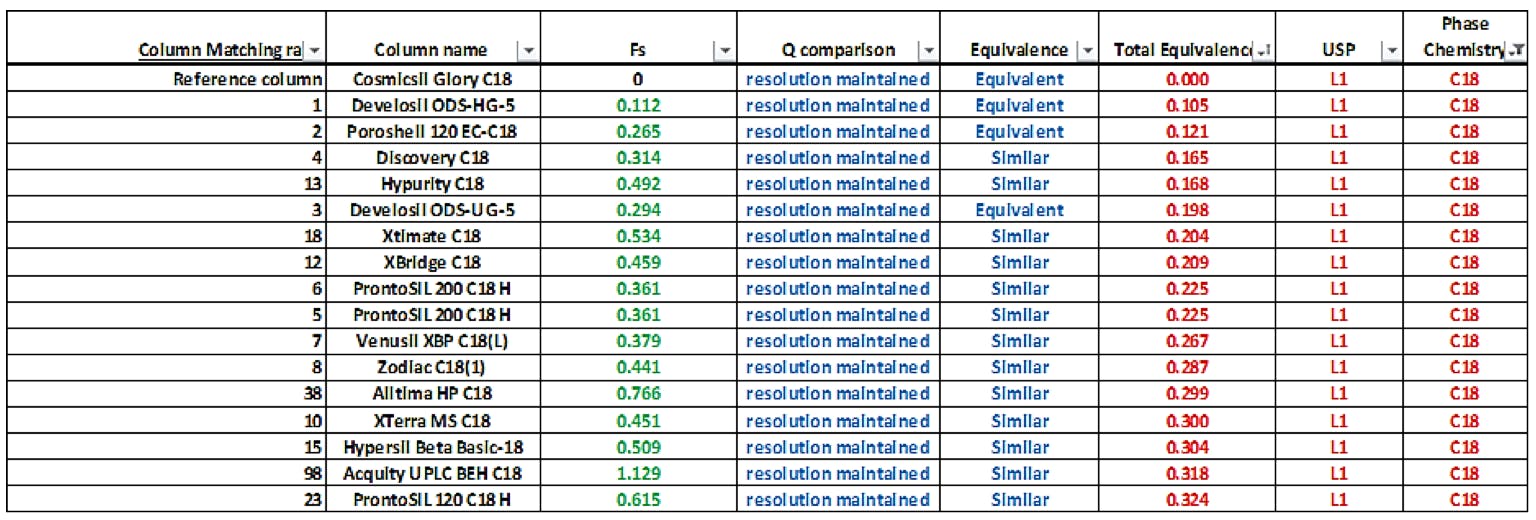
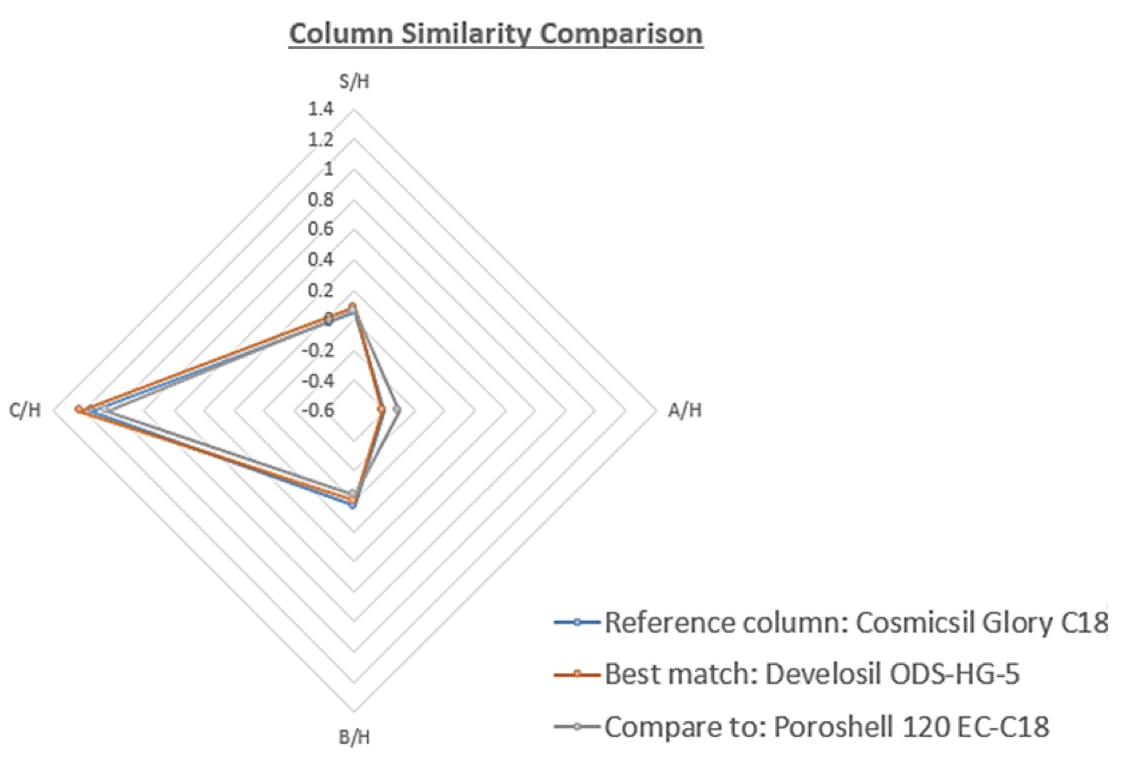
I would suggest that this approach would be far more applicable in defining the selectivity equivalence of columns and would be much less ambiguous (some might say more scientific) than the current proposed wording. However, maybe the intention is that the stationary phase cannot be changed. I would find this very restrictive in my own work, and I’m sure many of other chromatographers would agree.
We should also briefly mention the narrowing of the allowable adjustment to the absolute % of minor mobile phase constituents. In some ways the new proposed maximum variation of 2% absolute is more sensible and is welcomed as the previous allowable range could be too wide when the minor component is present at larger percentages in the isocratic mobile phase.
As an example;
50:50 Aq : Organic eluent - 30% change to either component would be 15% alteration of either aqueous or organic (35 - 65%A or B) so the 10% absolute limit applies and the allowable range is 40:60 or 60:40.
This would result in a large change in retention (by a factor of around x3!)
The new guidelines would limit this change to 48:52 or 52:48, as a worst case scenario, which I feel is much more appropriate.
It’s excellent to see that compendial authorities are considering updating ‘allowable change’ regulations, and the updates to allow changes to gradient profiles certainly open up a whole new world, but there are some reservations around the new restrictions on allowable changes to stationary phase chemistry.
References
- 1. N.S. Wilson, J.J. Gilroy, J.W. Dolan, L.R. Snyder, J. Chromatogr. A 1026 (2004) 91.
- 2. W. Melander, J. Stoveken, Cs. Horvath, J. Chromatogr. 199 (1980) 35.
- 3. J. Zhao, P.W. Carr, Anal. Chem. 70 (1998) 3619.
Related articles