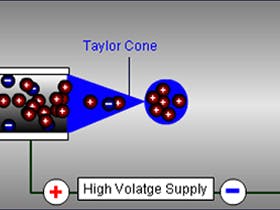
06 Jun 2022
Critical Evaluation of Analytical Methods: Liquid Chromatography with Mass Spectrometric Detection
By Tony Taylor
We continue our ‘Critical Evaluation’ series with a look at liquid chromatography methods which use mass spectrometric detection.
Concentrating on the detector settings provided in literature or method specifications for the mass spectrometric detector, we can learn about potential pitfalls before entering the laboratory.
Critically evaluating LC-MS detector settings is perhaps the most difficult of all the extra-laboratory detective skills to master. This is primarily due to the differences between instrument design and nomenclature used by various manufacturers for the variables associated with the source and analyser components. The recommended values and ranges for voltages, voltage offsets, gas flows, pressures and temperatures also make the critical evaluation of LC-MS detector settings particularly tricky. Perhaps this is why we, as a community of practitioners, tend to assume that many of the variables associated with our MS detectors for liquid chromatography as ‘lock and leave’ (that is, we set or optimise them once and then assume that these are applicable for all other applications). The notable exception here would be the capillary (sprayer) voltage, which tends to be considered the universal variable for optimisation in API interfaces. However, it could be argued that the differences between instrument designs and variables are an even bigger reason to be critical when adopting methods from scientific literature or instrument manufacturers' application notes, especially when the instrument used in the literature method differs from your own.
It's outside the scope of this article to delve into the theory of Atmospheric Pressure Ionisation (API) mass spectrometry, but for the interested reader, a short-form description of the relevant processes involved in electrospray mass spectrometry can be found in our previous blog: Optimising-LC-MS-sensitivity.
For all instruments, the following processes are critical to ionisation and ion sampling within electrospray LC-MS:
-
- Impart a charge to analyte molecule – typically through pH adjustment of the eluent or via induced charging or charge transfer in the electric filed at the tip of the capillary (sprayer)
- Accumulating an excess of positive or negative charge (depending upon detection mode) into a droplet at the tip of the capillary (sprayer)
- Increasing the charge (and hence the repulsive forces) at the surface of the sprayed droplet by solvent evaporation in order to induce droplet jet fission and achieve further charge density accumulation within the offspring droplet
- Liberating the charged analyte into the gas phase (several mechanisms are proposed)
- A preferential sampling of the charged gas-phase analytes into the vacuum region of the mass spectrometer
Within the mass analyser, further important processes which are common to all mass analysing devices include:
-
- Declustering and/or fragmenting the analyte prior to the (first) mass analysing device
- Filtering gas-phase ions based on their mass to charge ratio
- Fragmenting ions from the first mass analysing device (for MS/MS instruments) by accelerating the ions through a potential difference within a cell containing a collision gas at a given pressure or flow and temperature
The values for the variables given within LC-MS method specifications will all be associated with one of the processes highlighted above. The rest of the article will highlight which variable matches which process.
The method for our current study is for a multi-laboratory study of Δ9-tetrahydrocannabinol and its metabolites in whole blood extracts [1].
HPLC Parameters
Column: Bi-phenyl (100 x 2.1 mm)
Mobile phase A: 0.1% formic acid(aq)
Mobile phase B: 0.1% formic acid in acetonitrile
Eluent Flow Rate: 0.6 mL/min.
Gradient (%B): 55% for 3.5 mins, ballistic ramp to 80% B and hold for 2 minutes
MS Parameters
Mode: Positive electrospray ionization, dynamic multiple reaction monitoring
Gas Temperature: 320 oC
Gas Flow: 10 L/min [Nitrogen]
Nebulizer: 30 psi [Nitrogen]
Sheath Gas Temperature: 320 oC
Sheath Gas Flow: 12 L/min [Nitrogen]
Capillary Voltage: 3500 V
Cell Accelerator Voltage: 4 V
MRM Parameters
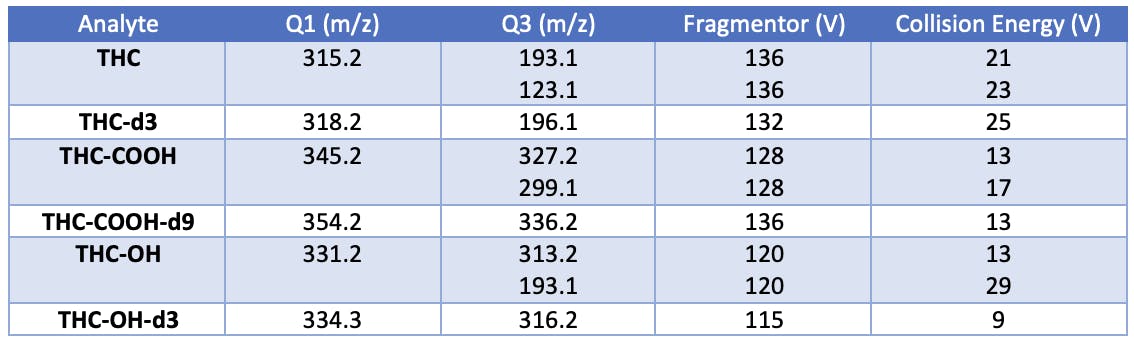
This is a particularly good literature method as it contains all of the information necessary to reproduce the method on a similar instrument or to make important decisions when transferring the method to a different manufacturer's instrument. This method wasn't used in anger, with no claims as to the validity or appropriateness of the settings for this particular application.
Considering the various settings, we can see what information the method might yield prior to donning laboratory PPE and getting hands-on with an instrument.
This is an electrospray API method (we’ll look at other ionisation modes in a later blog) and as such, it's important to ensure that the eluent flow into the MS interface is appropriate and does not interfere with droplet formation processes at the tip of the capillary (sprayer). If the droplets do not have time to form and accumulate charge prior to being dispelled from the tip then the instrument response will be poor or non-existent. In modern instruments, where nebulising gas is used to help constrain the droplet growth at the capillary tip in order to accumulate charged analyte, ‘usual’ eluent flow rates range from around 0.4 to 1.0 mL/min and anything outside of this range should be carefully considered and manufacturers literature consulted. It may be that nebulising gas flow rates can be adjusted to help with higher or lower eluent flows, but be prepared to make changes as necessary. At 0.6 mL/min. the eluent flow rate in this method seems to be appropriate.
As mentioned, the nebuliser gas pressure is used to control the flow of gas around the sprayer capillary and helps to ensure proper droplet formation at the capillary tip, which is essential for good sensitivity. The name of this variable and the appropriate setting and range vary between manufacturer and instrument design and it’s good practice to check instrument manuals to assess if the setting is to the very high or low end of the range in order to assess the validity of the setting. As noted above, it may be necessary to use more extreme values of nebuliser gas flow to cope with very high or very low eluent flow rates. From personal experience, when performing design of experiment (DoE) based source optimisation, the nebuliser gas flow rate doesn’t typically feature as a highly significant variable, and therefore this could be seen as a secondary variable when assessing the suitability of a method.
The capillary voltage is widely regarded as a (if not ‘the’) primary variable in affecting the efficiency and sensitivity of the electrospray process. Consider the API interface as an electrochemical cell which is completed in the gas phase. The voltage applied to this circuit controls several crucial processes, not least of which is the charge separation that occurs at the capillary (sprayer) tip. The processes are highly analyte and eluent dependent meaning that the voltage applied to the capillary is of crucial importance. The effective capillary voltage range for a particular instrument may be wider than you realise, even if the instrument is producing a signal, it may be far from optimal and may also produce unstable or erratic results. Where possible, it is always recommended that the applied voltage to the API is optimised for each application. 3500V seems like an appropriate value for the instrument cited in the application; however, be ready to optimise the applied voltage setting with each application and to take the view that this variable is not a one size fits all parameter. In DoE experiments, this variable is often identified as significant, but this is not always the case and it isn’t always the most significant variable. Consider the capillary (sprayer) voltage ‘in the round’ with the other interface parameters and don't attempt to optimise the experiment using this variable alone.
Drying gas temperature and flow (gas temperature & gas flow in the method conditions above) are used to control the desolvation of the electrospray droplets as they pass through the ion source. The desolvation process is crucial in accumulating surface charge density which leads to droplet jet fission and ultimately to the release of gas-phase analyte ions into the ion source. The droplet desolvation rate is controlled directly by the drying gas flow rate and temperature, as is the position within the inlet where the analyte ions are liberated into the gas phase. Gas-phase ion liberation needs to occur as close to the sampling ‘sweet spot’ as possible within the interface so that the applied voltage and vacuum can direct the charged analyte ions into the sampling orifice of the mass analysing device. Drying gas temperature and flow are often significant variables when optimising interface variables in electrospray ionisation experiments. Be be aware of the allowable range for these variables and assess where in the range the variables in the method under consideration are set. If the instrument used in the literature method is not the same as yours, then understanding where the variables are in the allowable range of the instrument used will help guide the setup of your own instrument.
The instrument used in the literature method uses sheath gas to form a temperature gradient concentrically around the capillary and within the ion source. This is purported to ‘focus’ the sprayed droplets, aid droplet desolvation and focus analyte ions into the ‘sweet spot’ of gas phase ion sampling within the ion source. In this case, sheath gas temperature and flow are likely to take precedent over the drying gas as primary source optimisation parameters. It’s also worth noting that other manufacturers have similar adaptations/improvements to the original electrospray source designs and each of these needs to be considered when assessing literature methods.
The fragmentor voltage controls the potential difference applied to the first vacuum region post ion source, which is under only partial vacuum due to the sampling orifice being open to atmospheric pressure in the ion source. Higher potential differences are used to accelerate analyte ions through this region in order to further desolvate analyte ions via collisions with the background air molecules and improve the ion filtering/selection process. At higher potential differences, and therefore rates of acceleration, analyte fragmentation can be induced, which can be a positive or a negative depending on the type of spectral experiment undertaken. In multiple reaction monitoring (MRM) experiments, specific precursor and product ions are selected by the first and second mass analyser in order to increase the specificity and sensitivity of the analysis. Take care that the fragmentor voltage is set correctly in order to ensure that the precursor ion selected for the first analyser is either correctly generated or is not destroyed in the first vacuum region. When the instrument used in the literature method is similar to yours, provided that the method states the required fragmentor voltages, then the instrument setup should be straightforward. However, if the instrument is different to yours, then ensure that the fragmentor (or equivalent) voltages are appropriate to ensure the optimum production (or preservation) of the precursor ion.
The collision cell voltage controls the acceleration of the precursor ion through this cell which is placed between the first and second mass analysers. Although in the high vacuum region of the instrument, the cell is filled with a collision gas (typically helium or argon) and accelerating ions through the gas will cause them to fragment through collision with the background gas, similar to the processes discussed in the fragmentor region. The higher the gas pressure (density) and the higher the collision cell voltage, the greater the degree of analyte fragmentation. Optimise the collision cell voltage in order to optimise the production of the analyte product ion and hence the sensitivity of the system. The collision cell energy (voltage) is analyte-specific and should be optimised for each analyte and application in the same way as described for the fragmentor voltage.
Several instruments provide a software-driven optimisation routine in which the fragmentor and collision energy voltages can be optimised either on an analyte by analyte basis or by taking into account the ‘universal’ response where standards of all analytes are not available. This point is important as, with all of the actions described above, one may need to assess the response of many target analytes, in which case a general optimum response may need to be considered, rather than having the luxury of optimising one or two target analytes for which pure standards are available for infusion or injection via the LC system.
Conclusion
These are some of the thinking processes to adopt when considering the use of a method obtained outside our own laboratory or organisation. A working knowledge of the instrument and principles of operation is very important in being able to do this effectively. Knowing the parameter ranges for each of the variables highlighted above is crucial in order to evaluate the method and sometimes this also requires us to know something about the instrument on which the original method was developed—to assess if the variable in question is at the higher or lower end of the range. We have only considered the major variables here, there will be many more associated with the tuning of the analysing devices for example, which weren't considered because they will be highly instrument specific. If your problem is of a more specific nature, all of our customers get direct access to our technical team. Get in touch to find out more.
The empirical truth is that one needs to pay careful attention to the optimisation of all of the ion source and mass analyser parameters mentioned in order to successfully supplant a method. Whilst we don't all have access to design of experiment approaches, a simple evaluation of the literature method against the settings used for other methods in our laboratory can help enormously with intercepting problems before entering the laboratory and troubleshooting them if and when they occur!
References
[1] https://www.oatext.com/multi-laboratory-validation-of-a-9-tetrahydrocannabinol-lc-ms-ms-test-kit-designed-for-quantifying-thc-and-marijuana-metabolites-in-blood.php
Related articles