03 Oct 2017
Hidden Problems in your LCMS data?
Hidden Problems in your LCMS data?
Take a look at the standard curve from an LC-MS determination of Alprazolam from a benzodiazepine screen. The peak area ratio of the analyte against its deuterated analogue is plotted against nominal concentration.
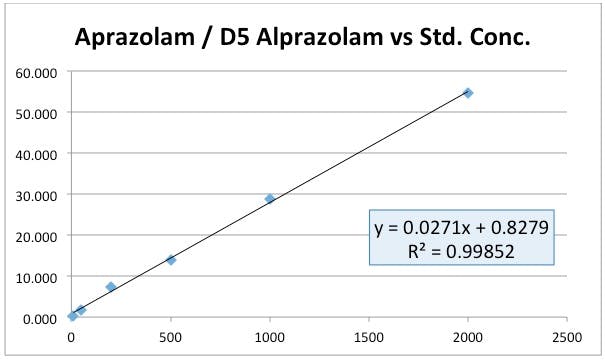
Figure 1: Calibration curve of Alprazolam / D5 Alprazolam peak area ratio against nominal standard concentration.
All good then – continue to collect or analyze the remainder of you data. Except the person acquiring data reported that there were issues with accuracy and reproducibility of QC samples at the lower concentrations and they had noticed that the theoretical ratio between analyte and internal standard did not match the ratio determined experimentally.
The raw data is shown in Table 1 and is represented graphically in Figures 2 &3.
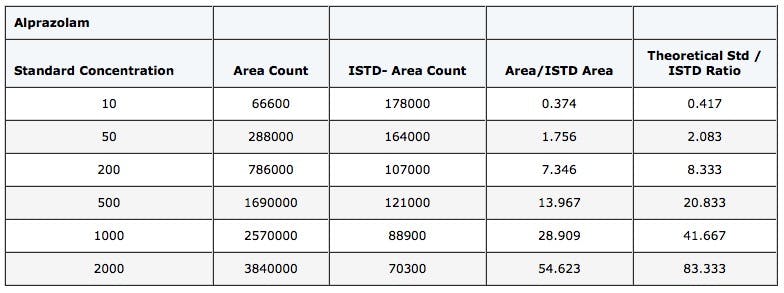
Table 1: Data from the ESI LCMS determination of Alprazolam using D5-Alprazolam as the internal standard.
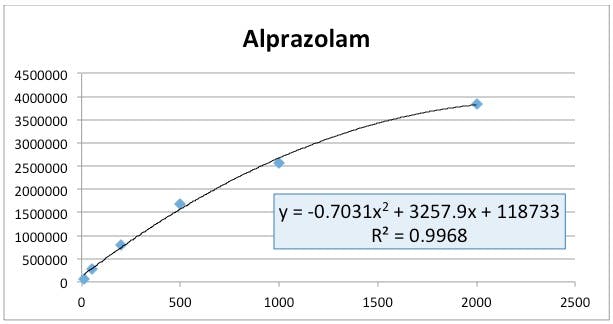
Figure 2: Alprazolam (analyte) peak area versus nominal standard concentration.
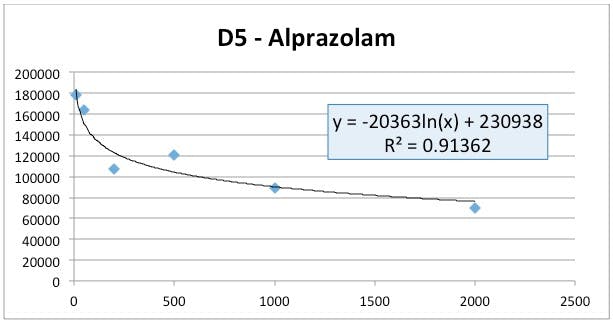
Figure 3: D5-Alprazolam (internal standard) peak area versus nominal standard concentration.
Notice anything?
Well – if this method was behaving properly, one would expect that the D5-Alprazolam would have an approximately constant peak area and it looks as though the area is decreasing logarithmically (and even then we just guessed at the best regression model!) (Figure 3). At the same time the alprazolam standard response looks to be following an approximately 2nd order polynomial response and higher concentration standard areas are clearly deviating from linearity (Figure 2).
So, as the alprazolam concentration increases the D5 Alprazolam area decreases and there seems to be some sort of issue with decreasing overall response with increasing analyte concentration.
This is a VERY COMMON phenomenon in LC-MS and if you don’t check your data for this type of issue then you may be missing something very important!
The issues which are causing the problem here are sometimes referred to as ‘ion suppression’ however the usual experiments carried out to investigate this phenomenon are unlikely to reveal the underlying cause.
Figure 4 shows an experiment in which the analyte is spiked into the HPLC eluent and a matrix blank (human plasma in this case) injected. If species within the matrix are artificially supressing (or enhancing) the analyte signal then a large negative or positive peak will occur as the matrix peak elutes into the detector; a typical example of this is shown in Figure 4, and indicates the matrix is suppressing the oxycodone signal.
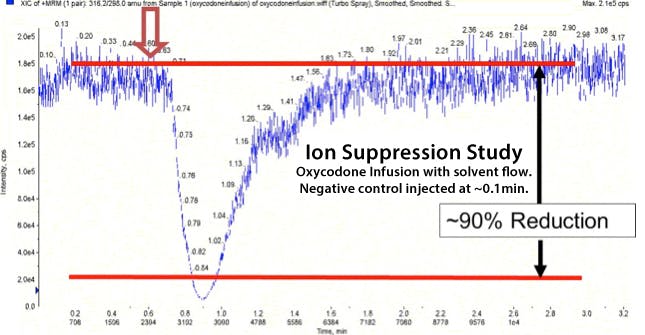
Figure 4: Resulting chromatogram from a Blank plasma matrix injection into an eluent spiked with easily detectable levels of Oxycodone.
In the case of our alprazolam experiment, this experiment caused virtually no baseline disturbance – indicating that the problem is not being caused by matrix interferences.
So what then? And how can we prevent the problem happening?
First a little theory about the nature of Electrospray Ionization – and I’ve tried to keep this as simple as possible, so the theory is a little ‘generalised’….
In ESI processes, droplets within the desolvation zone undergo ‘fissions’ in which some of the droplet volume and a lot of the droplet charges (analyte and other ions) leave the parent droplet through a series of these droplet fissions, eventually ending up in very low volume droplets (offspring droplets) with a lot of charge, from which the analyte ion can ‘evaporate’ into the gas and be samples into the mass analyser.
Analyte ions at the droplet surface have a better chance of making it into the offspring droplet and ultimately to evaporating into the gas phase – and therefore being detected.
The ability of an ion, surrounded by a cluster of water molecules, to make it from the bulk of the droplet to the surface of the droplet is a very important process and is influenced by the number charges on the molecule and its size (related to its mass) in solution. So if analytes have different mass and different numbers of charges (or polar functional groups) then they may move to the surface preferentially to other species and cause what we know as Ion Suppression (for more information see P. Kerbale and L. Tang. Anal. Chem., 65 (1993) 3654). However, this cannot explain the issue that we see here, because the analyte is ‘suppressing’ the internal standard which only differs in that it is deuterated.
What is VERY important is the fact that there are limited spaces at the droplet surface of either the parent or any offspring droplets. So – if any species is in great excess to another, by the laws of statistics, the high concentration species will be present at more of the surface sites, will make it into more offspring droplets and be there in higher concentration in the gas phase and hence in the mass analyser. Further, if the analyte is present in very high concentrations, there will come a point at which all surface sites are occupied and increasing the analyte concentration in the bulk will not increase the detector response.
This phenomenon is shown in Figure 5 – as the ammonium ion concentration is increases the signal due to the other ions reduces due to displacement from the droplet surface. We observe a limit to the signal intensity as the Alprazolam concentration is increased as all droplet surface sites are ‘saturated’ with analyte and as the analyte is present in the higher standards in vast excess to the deuterated analogue, the internal standard response will decrease with increasing analyte concentration.
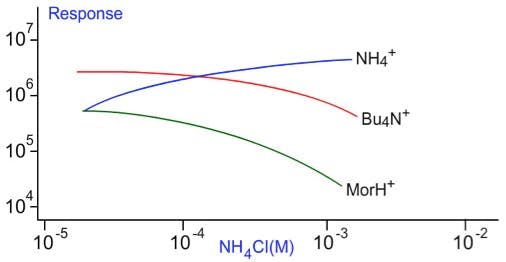
Figure 5: Tetrabutyl ammonium and protoned Morphine signals are suppressed by increasing ammonium ion concentration which also shows limiting behaviour in terms of signal intensity vs’ concentration (Mohamed S. Rashed, Minnie Jacob, Mohamed Al-Amoudi, Zuhair Rahbeeni, Moeen A.D. Al-Sayed, Lujane Al-Ahaidib, Amal A.A. Saadallah, and Sharon Legaspi1. “Rapid Determination of Orotic Acid in Urine by Liquid Chromatography–Electrospray Tandem Mass Spectrometry.” Clinical Chemistry 49, No. 3, 2003, Pp 499-501.
The good news is that there are certainly things that can be done to overcome our issues.
•Increase the concentration of the internal standard to match the concentration of the middle standard in the calibration curve to avoid ‘swamping’ the internal standard signal
•Dilute the sample by at least a factor of 10 in order to avoid the droplet surface becoming saturated with ions
•Reduce the nebulising gas flow rate – which allows each sprayed droplet to have a little more volume and therefore more surface area
•Adjust the drying gas temperature to adjust the position within the interface desolvation zone at which droplet fission begins and ultimately where the analytes reach the gas phase (optimum temperature may be higher or lower than the current value)
By experimenting with these parameters and techniques you will find an optimum combination to avoid suppression effects, the most powerful of which will be reducing the overall concentration of the sample injected.
My closing comment would be – if you don’t examine your LCMS data closely, you could be missing some of these effects and those problems with your quantitative data will never be fixed. Just saying!
Related articles