09 Jul 2018
Optimising Sensitivity for Splitless Capillary GC with FID Detection
|
It is often possible to achieve better sensitivity and lower limits of detection and quantitation using standard GC equipment – here I’m referring to a standard split / splitless injection port and a Flame Ionisation Detector (FID). Paying attention to some of the fundamental variables as well as some of the more esoteric considerations can lead to much improved method performance. Here we will consider all of the important aspects necessary to push the performance of standard capillary GC equipment to the limits. Whilst improving sample preparation may not be desirable in terms of analytical simplicity and throughput, it is certainly one aspect that can be used to increase the analyte concentration in the sample prior to injection. Selective techniques such as solid phase extraction (SPE) or dispersive solid phase extraction (dSPE) can be used to produce a final eluate which has the same concentration of analyte in a much smaller volume and this does not necessarily mean that the eluate needs to be evaporated – simply that the elution volumes used are smaller than the original sample volume and a good working knowledge of such techniques will allow this to be a routine consideration. The use of highly eluting solvents (in terms of organic strength, pH or organic strength), using multiple small aliquots of elution solvent which are allowed to ‘soak’ or remain stationary within the sorbent bed or even the use of supported liquid extraction (SLE) to replace traditional liquid / liquid extraction techniques are all good ways to improve the analyte concentration within the sample, prior to instrumental analysis. Other more advanced techniques such as solid phase micro-extraction (SPME), single drop micro-extraction (SDME) and dispersive liquid/liquid micro-extraction SPE (DSLLE) are all also available to help improve the analyte concentration so that standard GC equipment may be used to achieve the required limits of detection and quantification. |
 |
The following is a check list that can be used to ensure that all aspects of instrument analysis are optimised for sensitivity.
Sample Solvent - The selection of an appropriate sample solvent can help to improve sensitivity, especially by helping to improve the peak shape and hence optimise the signal to ratio achievable. Perhaps one of the least considered aspects of improving the sensitivity is that of improving the peak shape to improve the signal to noise ratio in terms of peak height. Gaussian peaks which are as narrow as possible (as highly efficient as possible) ensure that the peak height is optimum and therefore the signal to noise ratio is also optimised.
By choosing a sample solvent which matches as closely as possible the stationary phase polarity, the peak shape will be optimised with no tailing or broadening which might otherwise reduce the signal to noise ratio. Here we think of choosing non polar solvents such as n-hexane with non-polar stationary phase such as 1% polydimethyl siloxane (DB1, RTX-1 etc.) or methanol with polar stationary phases such as waxes.
Further, choosing sample solvents with lower expansion co-efficient will allow higher solvent (sample) volumes to be injected. This will be explained further in the next section.
High head pressure injection – the inlet liner is designed to hold a certain volume of gas (typically 500 – 950mL) and we need to avoid over filling the liner with sample gas or problems will occur with poor quantitative reproducibility and carry over. The volume of gas created by the sample injection will depend upon the inlet temperature, the inlet pressure (directly related to the carrier flow through the liner under splitless conditions) and the type and volume of sample solvent injected. By choosing a sample solvent with a lower expansion co-efficient and increasing the pressure within the liner during the injection to in order to constrain the volume of sample gas created, larger injection volumes can be made. Calculators, typically known as ‘backflash’ or ‘vapour volume’ calculators can be used to determine the maximum sample volume that can be injected before liner over-filling occurs.
For example, with an inlet temperature of 250oC, using a single taper 4mm i.d. splitless liner and a head pressure (inlet pressure) of 14psi, 2.3mL of hexane can be injected without compromising a ‘safety threshold’ limit of 75% of the liner volume filled with the resulting sample gas. If methanol is used under the same conditions, only 0.7mL of sample can be injected before the threshold limit is reached due to the much higher expansion coefficient of methanol versus hexane. If the head pressure of the inlet is increased to 60psi for a few seconds during the injection phase of the analysis, the volume of methanol which can be injected increases to 3.2mL which could be enough to achieve the desired sensitivity targets for the method. Incidentally, 10mL of hexane could be injected under these increased head pressure conditions without exceeding the threshold value!
Optimise Splitless Time – in splitless injection mode, the split vent vale is closed and all of the contents of the inlet are directed into the GC column. After a pre-determined length of time (the splitless time) the split vent value is opened and the remaining inlet contents are vented to atmosphere. This helps to prevent wide and badly tailing solvent peaks, increased baseline noise and a rising baseline during the analysis. If the split vent is opened too early, there is a risk that not all of the sample vapour has been transferred to the GC column and therefore sample (and sensitivity) will be lost. If the split vent is opened too early, the broad / tailing solvent peak may reduce the sensitivity of early eluting analytes and noisier baselines will reduce the signal to noise ratio for all analytes. One must experimentally determine the optimum splitless time in order to achieve the best reproducibility and sensitivity from the splitless injection.
Optimise the initial oven temperature and hold time – in splitless injection the analytes are introduced more slowly into the GC column and therefore analytes are spread into wide bands. It is important to re-focus each analyte as it enters the column so that peaks shapes are as narrow as possible. For early eluting analytes, this focussing occurs as the sample solvent condenses on the stationary phase and then slowly evaporates from the column entrance under the reduced vapour conditions created by the flowing carrier gas, pushing the analytes into an ever decreasing tight band of solvent. In order for the solvent condensation to occur, the initial oven temperature must be maintained at around 20oC below the boiling point of the sample solvent during the injection phase of the analysis. Further, to ensure a contiguous film on the inner wall of the column, the sample solvent polarity must be well matched to the polarity of the stationary phase.
Initial oven temperature hold – the focussing processes described above take a finite amount of time to occur and during this time the oven temperature must be held constant. However, if this initial hold time is too long, some degree of analyte dispersion may occur, reducing the peak efficiency and hence the signal to noise ratio. The initial hold time should be carefully evaluated if optimum sensitivity is to be achieved.
GC Column Choice - shorter columns (10-15m) with narrow i.d. (0.18 – 0.25mm) and thin films (<0.3μm) will give the best peak efficiencies and therefore the optimum signal to noise ratio. Further, less polar stationary phase chemistries will show less inherent bleed, again improving the signal to noise ratio, although the choice of stationary phase chemistry will be sample dependant. One should chose the least polar column with the thinnest film which results in a satisfactory separation, in order to reduce column bleed to the minimum amount possible.
Gas Quality – carrier gas quality will determine the amount of stationary phase bleed and therefore gas filters should be installed to remove moisture and oxygen. Hydrocarbon traps should be fitted to the FID gas supplies in order to reduce background noise. Use 99.999% pure gases in conjunction with gas filters where optimum sensitivity is required.
Column installation - make sure that column cuts are the best quality that they possibly can be and take great care to install the column properly into the inlet and the detector – taking special care to avoid dead volumes at the detector end which may lead to peak broadening and a subsequent reduction in signal to noise ratio.
Column contamination - if peak shape deterioration occurs and inlet causes cannot be identified as the cause of the problem, ensure that the column inlet is ‘trimmed’ to avoid peak shape deformation and maintain good peak shape and efficiency. Start with a 20-30cm column trim and work from there until the contaminated region of the column has been removed and peak shapes restored.
Carrier gas operating mode - ensure that the carrier gas operating mode is set to constant flow versus constant pressure to ensure that the carrier flows at the same linear velocity during the whole temperature program. This will avoid the carrier slowing as the temperature increases which can lead to broadening of later eluting peaks and sub optimal detector response.
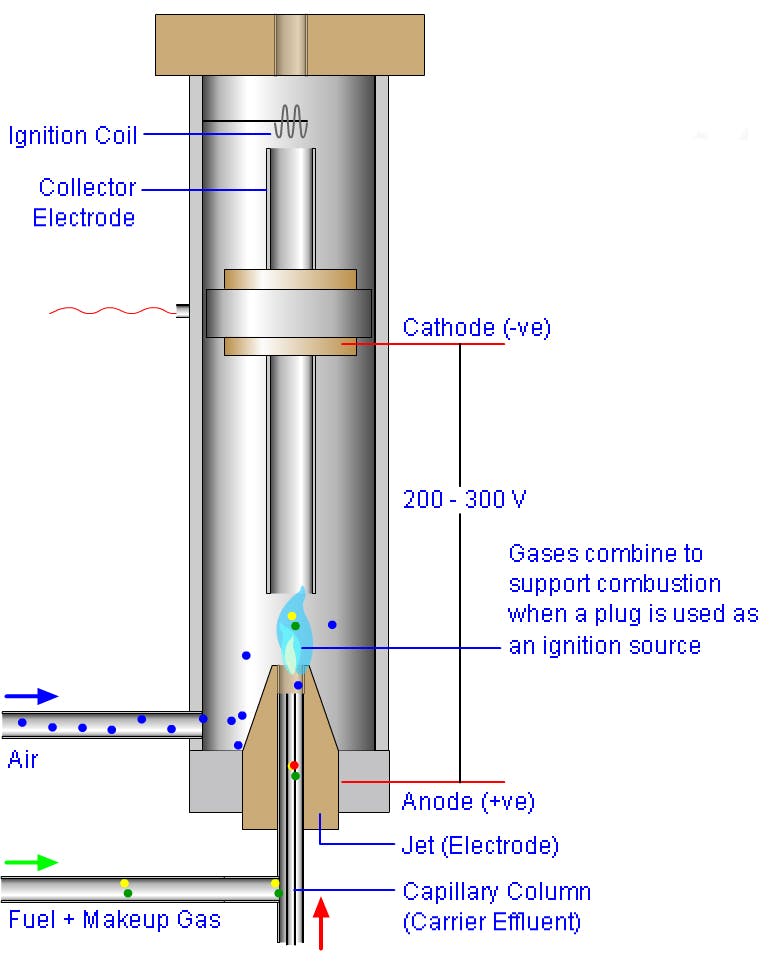
FID Optimisation - optimise the fuel (hydrogen) to oxidiser (air) ratio of the FID to ensure the best response for your analytes (typically start with a 10:1 ratio and adjust the fuel gas in steps of 10%. Also ensure that the make-up gas flow rate (Nitrogen is recommended as the most effective make-up gas by many manufacturers) is optimised and note that the make-up gas flow rate can have a pronounced effect on analyte sensitivity. Start with a 1:1 ratio of make-up gas to fuel gas and adjust in 10% steps to investigate the optimum range.
Thermal gradients - ballistic (short / fast) thermal gradients will produce the sharpest peaks and the best signal to noise ratios but ensure the oven heater motor can follow the required temperature profile in a reproducible way. Of course the use of ballistic thermal gradients will depend upon the separation requirements and the complexity of the sample.
Quality consumables - use the best quality septa and liners and inlet seals to ensure good peak shape, especially with polar analytes and ensure that the inlet is regularly maintained.
Whilst the above check list is long, with some discipline and good analytical practice, the limits of detection and quantitation of many GC methods can be markedly improved even when using ‘standard’ GC equipment of split/splitless inlet with flame ionisation detection.
Related articles