02 Oct 2017
Column Selection for HPLC Method Development
The approaches to method development and especially to column selection in HPLC vary widely and when I ask about column selection criteria I get a range of responses from:
- We use our favourite columns
- Our column screen has been developed for our applications types over a number of years
- We look for applications from literature and vendors using similar analytes
- We use what worked last time for a similar analyte / matrix
- We use whatever is on the instrument or the first column we put our hand on in the drawer
These responses range from very laissez-faire to seemingly well researched, but when pushed, even the most diligent of analysts fail to grasp the key factors in column screening.
The toolkit approach should provide a highly efficient and wide ranging exploration of the separation possibilities for the application under investigation, in particular the selectivity which will drive the separation under development.
Whilst it is true that a wide range of modifiers, pH’s, additives and gradient profiles are an important part of the selectivity ‘space’ it is also true that the nature of the stationary phase is a large contributor to selectivity – yet people are often a lot more ‘circumspect’ when discussing the factors which drive stationary phase choice.
It is particularly important to be aware of the characteristics of a stationary phase and it’s support which drive selectivity and a good set of descriptors can be found in the Hydrophobic Subtraction Model [1] of column selectivity which is used by the Product Quality Research Institute (PQRI) to build their useful database of column characteristics:
http://apps.usp.org/app/USPNF/columnsDB.html
This model can be very simply described using the graphic shown in Figure 1
Figure 1: Cartoon representation of five solute–column interactions of the Hydrophobic Subtraction Model.
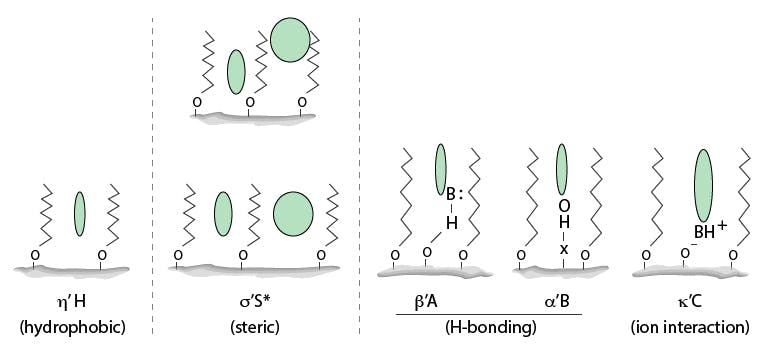
Figure 1: Cartoon representation of five solute–column interactions of the Hydrophobic Subtraction Model.
The terms beneath each picture in Figure 1 represent the terms which the Hydrophobic Subtraction Model defines as being of primary importance in determining separation selectivity behaviour of the analyte and column, which are determined using retention and selectivity measurements on each column using a defined set of chemical test probes.
The primary characteristics can be very simply described as:
H – is the hydrophobicity (retentivity) of the phase and is dictated by the type and bonding density of the ligand
S* - Steric selectivity, the ability to separate molecules whose shape, size and polar functional groups differ and is dictated by the type and bonding density of the ligand
A – the stationary phase acidity, represents the ability of the phase to hydrogen bond with basic functional groups of the analyte and a contributor is analyte interactions with acidic silanol groups on the silica substrate
B – the stationary phase basicity, represents the ability of the phase to hydrogen bond with analyte acidic functional groups which can be via surface metal ions or a layer of sorbed water at the silica surface
C – represents the ion exchange capacity of the column, which changes with increasing pH as the acidic silanol groups on the silica surface increase in charge between pH 2.8 and pH 7.0 (the two values at which the ion exchange capacity is measured in the model), thus affecting the degree of secondary interaction between polar and ionisable analyte functional groups and the silica surface
It’s important to highlight that each of the interactions above can contribute in a positive way to the selectivity of the separation.
The key to improving column selection, lies in understanding which characteristics dominate with each phase and what affects the characteristics have on the separation of certain (your!!) analytes. Before I go on to consider how this might be achieved, consider the number of times in the above list that the word silica appears versus the number of times I mention ‘ligand’. Whilst all of the key characteristics are affected by the bonded phase ligand, at least three of them are highly influenced by the nature and surface treatment of the underlying silica onto which the stationary phase ligands are bonded.
Given that, in order to properly explore the selectivity space available, one needs to be varying the support material as well as the bonded phase ligand in order to get the ‘full range’ of selectivity that might be available for a separation. So, if your screen contains a set of columns from a single manufacturer where the only difference is in the bonded phase ligand, you need to consider if you are fully exploring the column selectivity space available. Any nervousness about variability in the nature of the underlying silica can be almost wholly discounted in modern times as well established manufacturers have good control over any batch to batch variability in either the bonded phase or the silica substrate.
One way to visualise the differences between columns is to compare them using radar plots where the key differences and dominant characteristics can be displayed. One can then make some rudimentary empirical experiments to assess which of the characteristics is best at driving the separations which you perform. This may be a set of columns which show very subtle selectivity differences if you work on similar molecules and have identified the key characteristics for a successful separation or may be a column set with widely differing properties if you are running a general screen or perhaps where the analyte nature or structure is unknown.
Some typical radar plots are shown in Figures 2 to 4.
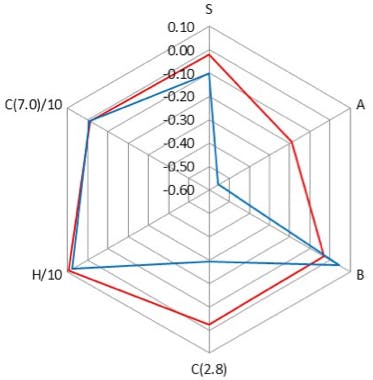
Figure 2: Comparison of properties of two different C18 (L1) HPLC column stationary phases using a radar plot of the PQRI column classification properties which are in turn based on the terms of the Hydrophobic Subtraction Model.
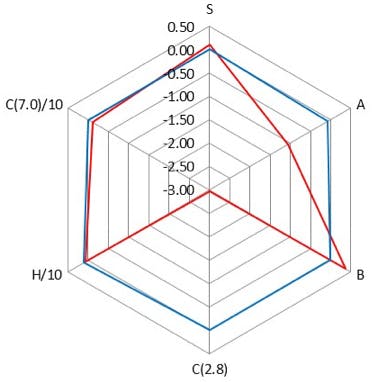
Figure 3: Comparison of properties of a Polar Embedded C18 column (L60) and a C18 column (L1) stationary phases using the radar plot of the PQRI column classification.
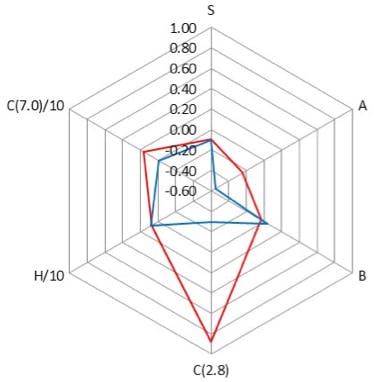
Figure 4: Comparison of properties of a PentaFluoroPropyl (PFP) column (L43) and a Phenyl column (L11) stationary phases using the radar plot of the PQRI column classification.
One should note the scales on Figures 2, 3 and 4.
Figure 2 shows subtle differences (full scale of radar plot is 0.35 units) in Steric (S), Ion Exchange (silanol acidity) (C2.8) and Surface Acidity (A) between the two C18 phases. However very large differences (full scale of radar plot is 3.5 units) between Ion Exchange capacity, Surface Acidity and Surface Basicity (B) are highlighted between the C18 and Polar Embedded columns compared in Figure 3.
Figure 4 compares two columns whose selectivity differs only in the bonded phase as the underlying silica used is identical. One column contains an aromatic system which is highly electron rich (Phenyl) and the other an aromatic system which is highly electron deficient (PFP) and as such the characteristics are markedly different (full scale of radar plot is 1.6 units).
The differences between the phases in Figure 2 are driven primarily by the underlying silica and bonding density changes and in Figure 3 by a mixture of the differences in the bonded phase as well as the differences in the underlying silica. The differences in selectivity between the columns in Figure 4 are predominantly due to differences in the bonded phase.
The key to good column selection is knowing the dominant characteristics required from the chosen column set which drive the separation of your analyte types, or covering the widest selectivity range possible if screening unknown or unfamiliar analytes.
The use of in-silico models such as the PQRI database and a better understanding of selectivity models such as the Hydrophobic Subtraction Model as well as an empirical appreciation of the correlation between column properties and various analyte structural features can significantly improve column selection for one off method development and as part of screening databases.
If you’d like to know more about your column sets or help with choosing orthogonal phases for your screen or a particular method development then please contact me at [email protected]
[1] N.S. Wilson, M.D. Nelson, J.W. Dolan, L.R. Snyder, R.G. Wolcott, and P.W. Carr, J. Chromatogr. A 961, 195–215 (2002).
Related articles