11 Jun 2018
Understanding stationary phases for HILIC separations
I spoke at a conference a few years ago and was describing our use of mixed mode chromatography and the difficult problems it had helped us to solve. There were howls from the audience when I described the number and type of variables that need to be considered (and optimised) when using mixed mode chromatography columns, the complaints mainly being centred around the complexity and how could these methods be robust and we already have a enough variables to consider and we don’t need any more selectivity etc. etc.
HILIC chromatography involves not only hydrophilic partitioning but potentially also hydrogen bonding, ion exchange (analyte / stationary phase), more ion exchange (salt additives / stationary phase), ionic (charge) repulsion and ion exclusion (displacement) mechanisms. What’s more, depending upon the stationary phase used, partitioning may not be the dominant retention mechanism.
My point? HILIC chromatography is not straightforward and there may be a number of mechanisms in play which need to be considered. Yet there were no complaints from the same audience who had complained so bitterly at the complexity of mixed mode chromatography when I recently lectured on some of our work on HILIC separations using a variety of different stationary phases. Breaking news – HILIC is mixed mode chromatography, at least some of the time!
I wrote some practical tips on the use of HILIC chromatography in this column in July 2017 and I wanted to expand on some of those thoughts, especially when it comes to understanding the nature of the stationary phases and their interactions with analytes and mobile phase constituents. Without a basic understanding of HILIC stationary phases and the retention mechanisms in play, it is very difficult to understand this type of separation, what to do when things go wrong or how to develop and optimise methods.
Let’s take a simple amino stationary phase as an example and examine some of the interactions which might be in play during a HILIC separation involving amino benzoic acid.

Figure 1: Schematic Representation of the HILIC Retention of 4-amino benzoic acid on an amino stationary phase.
There are many possible contributions to retention from this very simple stationary phase and analyte combinations;
1) The hydrophilic nature of the stationary phase will dictate (amongst other factors) the thickness of the adsorbed water layer at the silica surface. This is turn will dictate the retention by partitioning from the eluent into the water enriched layer for more polar (hydrophilic) analytes and analyte moieties (functional groups).
2) The thickness of this water layer may be affected by altering the % organic within the eluent
3) Depending upon the eluent pH the residual silanol groups on the silica surface may be charged (typically < pH4) and therefore ion exchange may occur between analyte cationic groups
4) If the analyte contains anionic groups, then ion repulsion may occur between the analyte and charged silanol groups on the silica surface, again dependent upon eluent pH. Similarly, if the analyte is cationic there may be electrostatic repulsion between the bonded phase ligand and the analyte.
5) If either the silanol group or the analyte are not charged, then the presence of polar analyte functional groups may allow hydrogen bonding between the analyte and silica surface. Further, bonded phases containing polar neutral groups may be able to hydrogen bond with neutral polar analyte functional groups and this type of interaction can play a major role in HILIC separations.
6) Under the pH conditions shown, the stationary phase is positively charged and the analyte is negatively charged – resulting in electrostatic interactions
7) If the eluent contains salts (ammonium acetate for example), then depending upon the type and concentration of ions, the analyte electrostatic interactions may be overcome by increasing the concentration or counter ion strength (position within the Hofmeister series)
8) Van der Waals interactions will also be in play between the hydrophobic portions of the stationary phase and the analyte
So to summarise, with this simple analyte / stationary phase combination, the following interactions / mechanisms of interaction will be possible:
- Hydrophilic partitioning
- Ion exchange
- Ion repulsion
- Hydrogen Bonding
- Ion Exclusion (depending upon the nature and concentration of eluent buffers)
- Van der Waals (hydrophobic) interactions between the analyte and stationary phase bonded ligand
Therefore, a mixed mode (some might say multi-mode) separation is in play and if all of the possible interactions are not understood and accounted for, then retention and selectivity in HILIC mode will be difficult to predict or influence using experimental variables.
The possibility and extent of these interactions will depend upon several experimental variables and will change with the nature of the stationary phase selected. Some stationary phases are only capable of some of the potential interactions and this will be explained in more detail later.
First a brief summary of the experimental variables in HILIC chromatography and how they might be altered;
Hydrophilic Partitioning – the strength of hydrophilic partitioning into the water enriched layer is influenced by the stationary phase chosen and the amount of organic within the eluent. Higher organic will result in stronger partitioning of polar (hydrophilic) analytes into the water enriched layer and therefore retention will increase. The eluent salt (buffer) concentration may also affect the thickness of the water layer.
Electrostatic Interactions – will depend upon the degree of ionisation of;
- The analyte (ionogenic or non-ionogenic)
- The bonded phase (if ionogenic)
- The Silica Surface (residual silanol groups)
To manipulate a HILIC separation using pH, we would need to know the solution pH and the analyte pKa value in order to estimate the degree of charge of the analyte. It is especially important to note that under the highly organic conditions that are typical of HILIC separations, both the analyte pKa and solution pH will differ from literature values or from the measured pH of the aqueous phase only. Some very approximate rules of thumb can be employed to help us assess the solution pH and degree of analyte ionisation;
| % Acetonitrile in Eluent | Target pH | Shift from aqueous pH |
| 90 | 2 | +3.5 |
| 90 | 4 | +2.5 |
| 90 | 6 | +2.0 |
| 75 | 2 | +2.5 |
| 75 | 4 | +2.0 |
| 75 | 6 | +1.5 |
| 50 | 2 | +1.5 |
| 50 | 4 | +1.0 |
| 50 | 6 | +0.5 |
| % Acetonitrile in Eluent | Shift from Apparent Analyte pKa (Basic Analytes) |
| 90 | -1.0 |
| 75 | -0.6 |
| 50 | -0.1 |
Table 1 & 2: Adjustments to eluent pH and analyte pKa in highly organic eluents.
So we must take care when planning a HILIC separation involving ionogenic analytes or interpreting chromatography, to adjust our thinking on the degree of analyte ionisation according to the amount of acetonitrile within the eluent, the literature value of the analyte pKa and the target eluent pH.
Some stationary phases are designed to carry a full charge in solution (permanently ionised) and others, such as the zwitterionic phases, carry both positive and negative charges to reduce the strength of electrostatic interactions and the these phases carry smaller net negative or positive charges depending upon the terminal group of the bonded phase. We will discuss the various stationary phases and their ability to interact electrostatically below, however we should note that with some ligands such as the pentafluoropropyl (PFP) stationary phase, retention is almost totally dominated by the strength of electrostatic interactions with analytes and that very little hydrophilic partitioning is involved.
Of course, when considering the electrostatic interactions between analyte and bonded phase, we should not forget that there may also be repulsion between the analyte and the bonded phase where both are either anionic or cationic. These repulsive forces will act to reduce retention and alter selectivity between ionogenic and non-ionogenic analytes.
The silica surface will have a pKa of around 4, so increasing eluent pH above 4 will make the silica surface increasingly anionic and therefore electrostatic attraction or repulsion also need to be taken into account when interpreting HILIC separations.
Hydrogen Bonding – polar analytes and stationary phase moieties may be able to hydrogen bond in order to increase retention and alter the relative selectivity of the separation between polar and less-polar analytes depending upon the degree of hydrogen bonding.
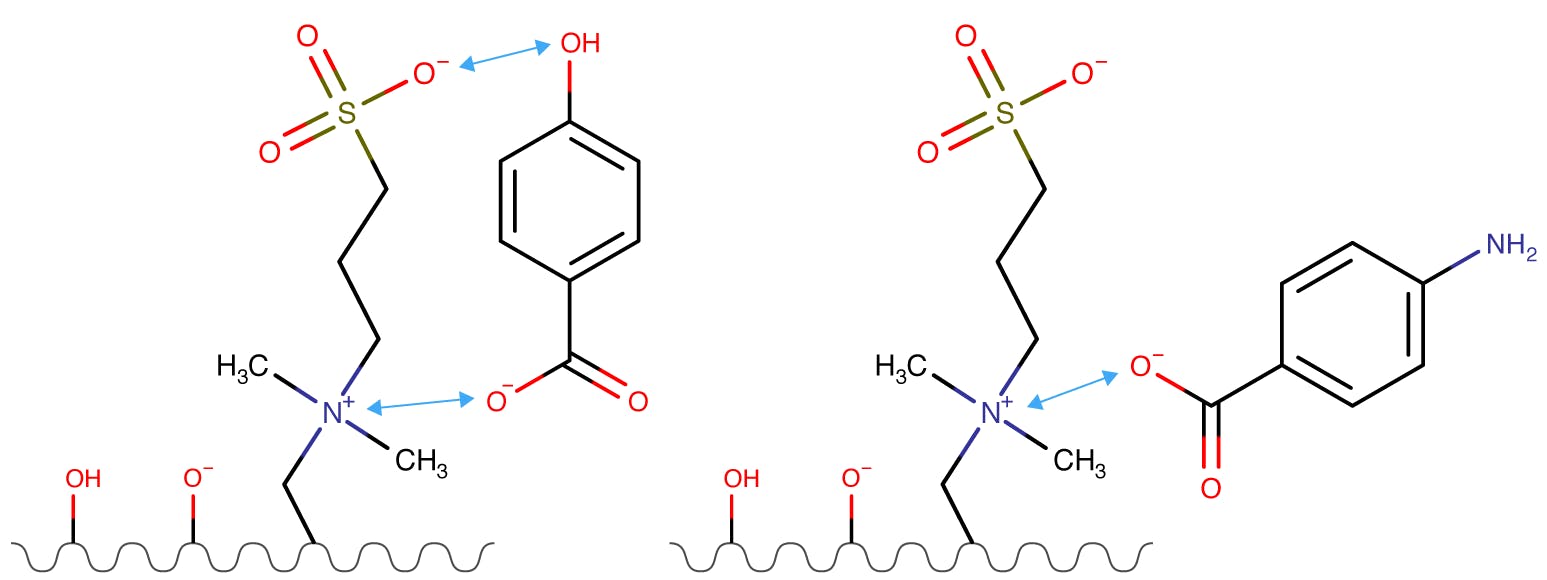
Figure 2: Interactions of 4 hydroxy benzoic acid and 4 amiono benzoic acid (right) with a zwitterionic stationary phase.
Figure 2 shows 4-hydroxy aminobenzoic acid (left) undergoing both electrostatic and hydrogen bonding interactions with the zwitterionic stationary phase. Hydrogen bonding of 4-amino benzoic acid is not possible and therefore the retention time of this analyte may expected to be shorter. It should be noted that both analytes will undergo hydrophilic partitioning and electrostatic interactions and therefore the effects of increased or reduced hydrogen bonding may be less significant.
The effects of hydrogen bonding in HILIC separations can be critical when separating polar neutral analytes.
Ion Exclusion (use of buffers) – typically the type and concentration of salt used in the eluent system will affect the degree of electrostatic interactions between the analyte and the stationary phase. For some separations, altering the buffer between a formate and acetate system can have a profound effect on the selectivity of the separation. Similarly, the concentration of the buffer will attenuate the degree of electrostatic interactions and salt concentrations between 5mM and 100mM are typical. Higher buffer salt concentrations tend to lead to shorter retention times when electrostatic retention mechanisms are in play. It should be noted that a good starting point for investigations into the effects of buffer concentration is around 20mM but if using eluents containing 95% organic, the buffer concentration should be kept below 15mM to avoid buffer precipitation.
It is also important to note that the amount of salt can also affect the thickness of the water layer and so can alter retention when dealing with neutral analytes. Higher buffer concentrations can lead to thicker water enriched layers and therefore longer retention times for neutral analytes.
Let’s conclude by considering some of the more popular stationary phase types and the types of interaction / retention mechanisms we can expect;
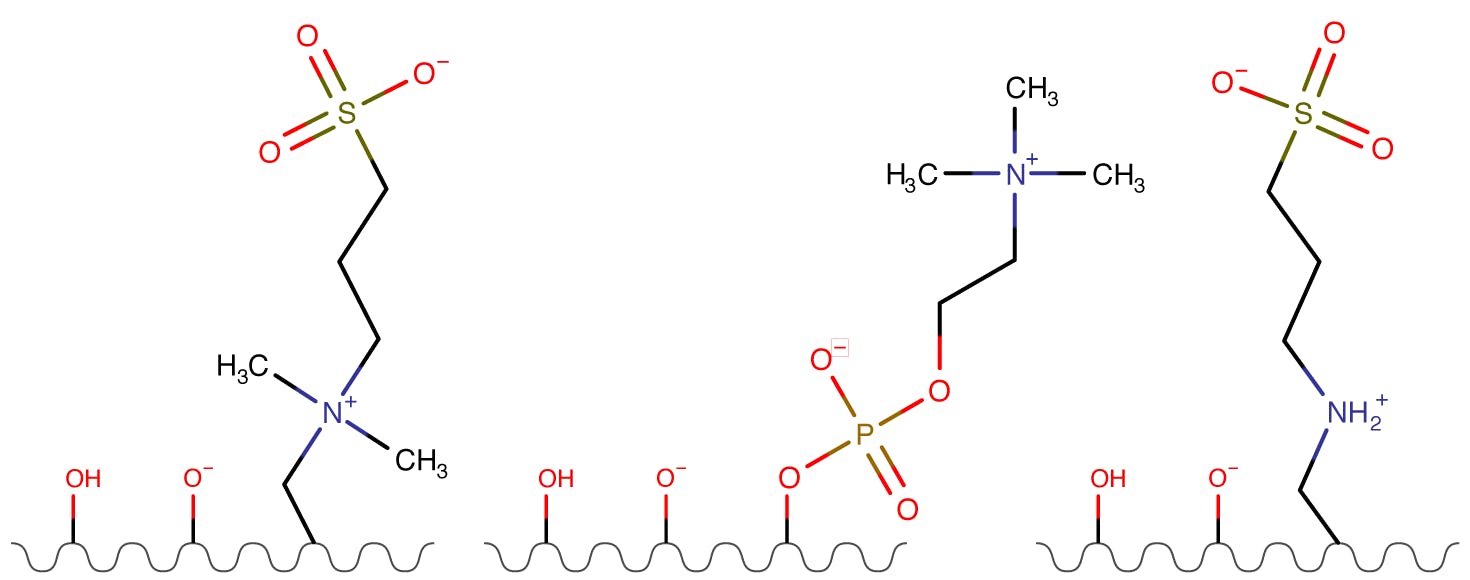
Figure 3: Zwitterionic Stationary Phases.
| Partitioning | Medium |
| Ion Exchange | Medium |
| Hydrogen Bonding | High |
These phases tend to have thick water layers and will undergo electrostatic interactions. The charge tends to be relatively low as the 1:1 ratio of positive and negative groups on each ligand will somewhat balance, the overall net charge being dictated by the terminal group on each phase (so net negative when the sulphonic acid group is terminal on the ligand for example). The overall extent of charge will not be affected by the solution pH and this can often simplify the planning and interpretation of separations, where the pH affects only the degree of ionisation of any ionogenic analytes.
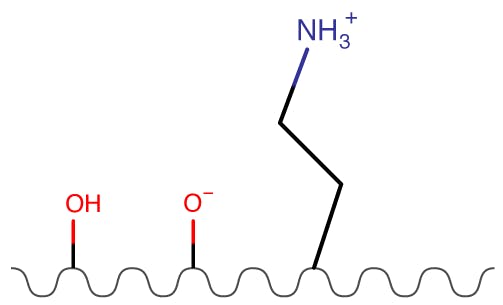
Figure 4: Amino Phases.
| Partitioning | High |
| Ion Exchange | High |
| Hydrogen Bonding | Medium |
Amino phases are charged and the degree of charge can be influenced by eluent pH. They are particularly retentive for acidic compounds but often suffer from increased equilibration times. Care should be taken when dealing with aldehydes as there is the possibility of the formation of Schiff bases with the stationary phase.
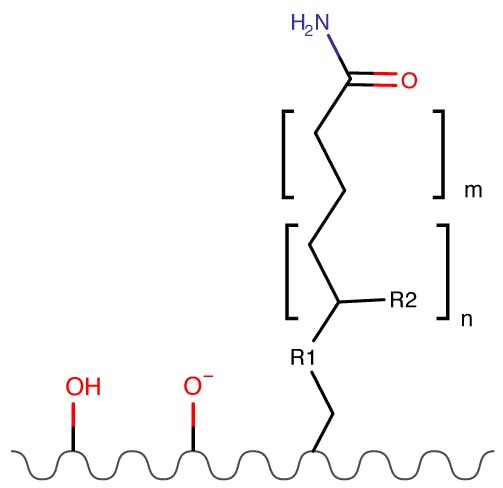
Figure 5: Amide Phases.
| Partitioning | High |
| Ion Exchange | None |
| Hydrogen Bonding | Strong |
In most commercial phases the alkyl spacer length is short. These phases are not ionisable and therefore the retention mechanism is easier to understand. Retention and selectivity can be controlled by lower salt concentrations.
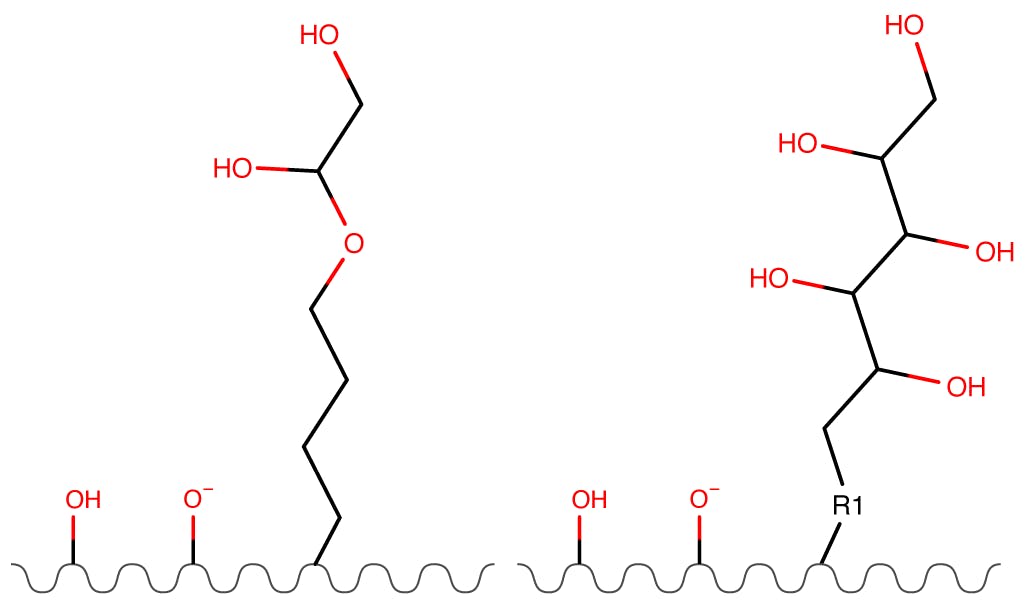
Figure 6: Diol and Polyol Phases.
| Partitioning | High |
| Ion Exchange | None |
| Hydrogen Bonding | High |
These phase are neutral and tend to have lower hydrophilic partitioning (more shallow water layers). The degree of hydrogen bonding is critical for these phases.
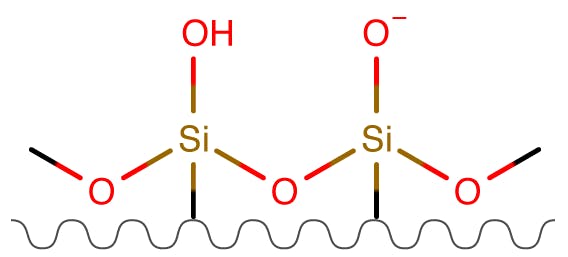
Figure 7: Bare Silica Phases.
| Partitioning | Medium |
| Ion Exchange | High (>pH4-5) |
| Hydrogen Bonding | Medium |
Whilst bare silica was popular in the early days of HILIC separations, silica surfaces are less homogenous and the pKa of the silanol groups can vary depending upon the metal ion content and surface treatments. For these reasons, bare silica is less popular now that dedicated bonded phases are available.
Figure 8: PentaFluoro Propyl (PFP) Phases.
| Partitioning | Low |
| Ion Exchange | High |
| Hydrogen Bonding | High |
These phase separate almost solely on the strength of electrostatic interactions between the analyte and stationary phase, with very little hydrophobic partitioning.
Whilst there are a number of other, more specialist phases available, the above treatment covers the most popular phase types and give a flavour of the multiple retention mechanisms possible within the HILIC separation mode. Without a good understanding of the stationary phase and its possible interactions, it is very difficult to interpret or plan effective HILIC separations.
The more eagle eyed reader will note that I have omitted the Cyano phase from the list above. In our experience their lack of hydrogen bonding capability and often poor retention of polar analytes even at high organic concentrations makes them less favourable for HILIC separations.
Related articles