03 Oct 2017
Retention Time Variability in HPLC
I’m sure we have all experienced it – that sinking feeling when you realise your analyte retention times have drifted outside the software ‘window’ and you have a pile of chromatograms with no quantitative results. Or you are trying to get that system suitability result to begin your batch of analysis as you really need to get out of the door fast but your retention times just won’t settle down. Or you are trying to reproduce Bob from the R&D centres’ method and his retention times (or chromatogram…..!) look nothing like yours. Or you are trying to validate your method and the three column lots you are trying just give different retention behaviour to the column that you just developed your method on. Or every time you do an injection the retention time of the analyte changes just a little – doesn’t cause anything to fall over, but you just don’t understand why.
Yes – there’s a whole bunch of retention time issues that cause us problems in HPLC. A lot of the underlying causes we can do something about – others we just need to be aware of the cause and put our minds to rest. The remainder of this technical tip will outline how to overcome, or better control, all of the situations outlined above.
Let’s start with retention time drift. This is typically due to a change in mobile phase composition, which can be caused when pre-mixed mobile phases lose organic through evaporation as the run progresses. Ever noticed this seems to happen more towards the end of a run? Well of course the organic is being continually lost to the atmosphere – or as the eluent in the sealed bottle depletes there is more headspace for the more volatile component to evaporate into - and of course a small amount of evaporation makes a bigger overall composition change in the ever diminishing volume of liquid! This is typically why we see elution times becoming longer rather than shorter. What to do? Mix eluents (even isocratic ones) online or at the very least ensure the reservoir you are using is capped. We may also experience a change in the pH of the aqueous component of the eluent over time caused by ingress of CO2 – lowering the eluent pH and changing the retention and perhaps even the selectivity of the separation….so again, cap your bottle. Do not use lab film to cover eluent reservoirs – especially when using MS detection (watch out for ions at 142 Da as you leach the plasticiser from the film!)
Another related note on eluents here – if we de-gas pre-mixed mobile phases using vacuum, the very act of sucking the mobile phase through the filter under vacuum can cause loss of the more volatile component – which will obviously lead to irreproducible changes in eluent composition from batch to batch of eluent. The same is also true when degassing pre-mixed phases using ultrasonic baths – the warming of the eluent in the sonic bath can lead to loss of the organic modifier and hence change retention characteristics!
Of course temperature is another variable that can alter retention time, changing not only the viscosity of the eluent but the kinetics of the retention mechanism, and ionisable compounds tend to be affected by temperature more than non-ionogenic compounds – so selectivity may also change. Most systems come with column heaters / chillers these days, but if yours doesn’t, and you get large temperature variations in the lab, this can cause retention time variability (especially when the system is placed directly below your air-con!). Even systems which do have column heaters work in different ways – some pre-warm the eluent prior to entry into the column for example and these systems may well give different retention times to those which heat the column only!
Equilibrating or priming a column when beginning an analysis can also show up some strange retention time shifts and variability. Without going into too much detail, this is due to the stationary phase surface being modified by your eluent or sample components. Primarily it’s the ‘wetting’ of the surface (especially with more hydrophobic phases such as C18) as the bonded phase takes on a layer of hydration - a slightly crass description but one which will do for this short tip. Further – the polar or ionised silanol (Si-OH) groups on the silica surface can irreversibly bind with polar analyte components or buffer ions to change the overall surface polarity. What to do – well you can try to inject a 10x more concentrated sample than you normally would to try and achieve the equilibration is a shorter time (fewer injections)!
And what of the situation in which the column you used to develop your method doesn’t behave like the new shiny ones you just bought to do your validation? Same as the previous situation really. Everything you put down the column (eluent and samples) modifies the surface – sometimes irreversibly, and the same goes for all of the stuff your colleague also put down the column before you used it to develop your method. What to do – buy a new column for method development and let it equilibrate properly before using it. If you have an ion pair reagent (and remember TFA is an ion pair reagent) and you remove it, use a different pairing reagent or switch to a different eluent modifier – you should contact your column supplier before continuing with method development – it may well be that you need a new column!
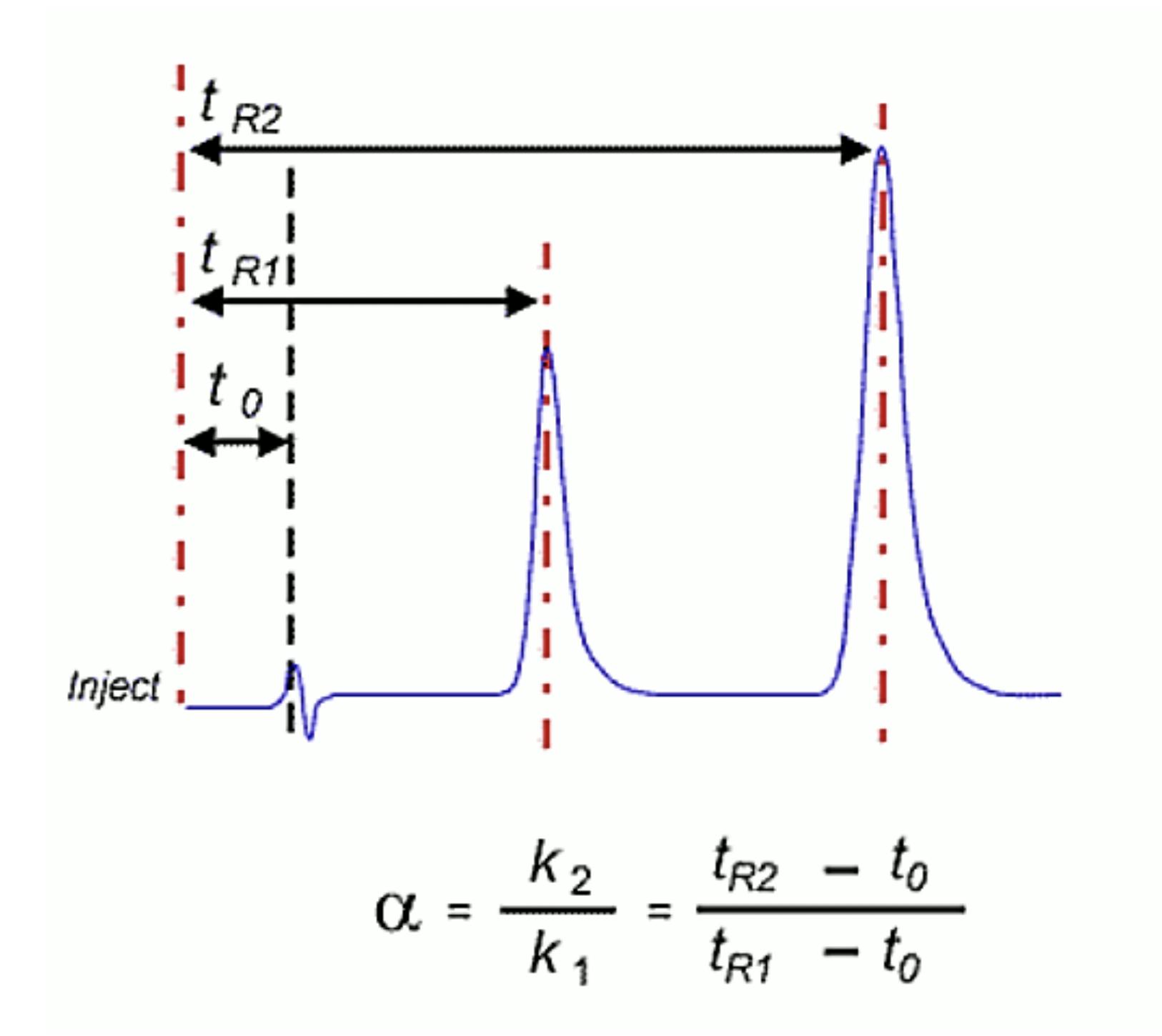
Matching retention times with Bob from R&D’s method is another tricky situation. You must make sure that you are preparing the eluent in the same way – including weighing solid buffers, taking care with volumetric work, adding organic to aqueous portions and adjusting the pH using the same acid or base and doing so with a properly calibrated pH meter. Further, the use of the same buffer is important – and just a note to all users of phosphate buffers – monosodium dihydrogen orthophosphate is not the same as disodium monohydrogen orthophosphate and neither of them has real buffering capacity between pH 3 and 6! Crucially, if the method involves gradient elution, you REALLY MUST know the gradient dwell volume for each system and adjust for any differences prior to repeating the method. If Bob’s dwell volume is shorter than yours then you need to use an instrument capable of injection delay (injection occurring after the gradient has started) and if his dwell volume is larger than yours, add an isocratic hold at the start of the method equal to the difference in volume x flow rate. If he was smart when he developed the method, then he would have inserted an isocratic portion at the start of the gradient which you can adjust to make sure the gradient dwell volume differences are catered for.
Pumps pump at a fixed flow rate – until they leak or break! Now most times poor pump performance will be accompanied by other symptoms such as low, high or cycling back pressure – but sometimes not. The easiest way to check your flow rate accuracy is to run the eluent into a 10mL volumetric cylinder and time it. If you get 10 mL in 10 mins when operating at 1 mL/min. then all is well – if not – all is bad and you need to get the system checked out.
Lastly, the more esoteric issue of sample diluent. For reasons that are too detailed to enter into here, the eluotropic strength and ionic strength of the sample diluent can sometimes have a big effect on analyte retention time and peak shape – yes that’s the sample diluent, the thing you dissolve the sample in – not the HPLC eluent. You should always strive to match the aqueous / organic ratio of the eluent (at the start of the gradient if doing gradient elution) as well as the buffer strength of the eluent . If you need to make the diluent more highly organic than the eluent (for solubility reasons) – try to restrict your injection volume to 10µl or less.
Related articles